Case Report
Monoclonal Gammopathy in a Pediatric Patient with Ataxia-Telangectasia: A Case Report, Review of the Literature, and Preliminary Differential Diagnosis
Jones TE1*, Shurin MR2 and Wheeler SE2
1Department of Pathology, School of Medicine, University of Pittsburgh, Pittsburgh, PA, USA
2Division of Clinical Immunopathology, Department of Pathology, University of Pittsburgh Medical Center, Pittsburgh, PA, USA
*Corresponding author: Sarah Wheeler, Division of Clinical Immunopathology, University of Pittsburgh Medical Center, Clinical Lab Building, Room 4024, 3477 Euler Way, Pittsburgh
Published: 28 Jun, 2018
Cite this article as: Jones TE, Shurin MR, Wheeler SE.
Monoclonal Gammopathy in a Pediatric
Patient with Ataxia-Telangectasia: A
Case Report, Review of the Literature,
and Preliminary Differential Diagnosis.
Ann Clin Case Rep. 2018; 3: 1528.
Abstract
Ataxia-Telangiectasia (A-T) is an autosomal recessive disorder characterized by immunodeficiency and neurodegeneration. An additional consequence of mutations in the ATM gene is a predisposition to monoclonal and oligoclonal gammopathies, which are reported in 8% of A-T patients. They have been hypothesized to originate from exposure of lymphocytes to events causing double stranded DNA breaks, such as ionizing radiation. Persistence of these breaks, along with the abnormal thymic development and defective cell cycle regulation seen in A-T, has the potential to lead to clonal dysregulation of B cells and to gammopathies. Of gammopathies present in the pediatric population, etiologies vary from autoimmune disorders, hematologic malignancies, myelodysplasias, and renal and hepatic disorders. Herein we discuss the unusual case of a pediatric patient with A-T, IgA deficiency, and asthma, who was found to have a monoclonal gammopathy. Further studies did not reveal the presence of an underlying malignancy or autoimmune disorder but the patient will continue to be closely monitored.
Introduction
Ataxia-Telangiectasia (A-T) is an autosomal recessive disorder characterized by
immunodeficiency and neurodegeneration. The disorder is caused by mutations in the
Ataxia Telangiectasia Mutated (ATM) gene on chromosome 11q22-23, which encodes a
Phosphatidylinositol 3-Kinase (PI3-K) involved in regulation of cell death, the cell cycle, DNA
repair and maintenance, and immune gene recombination [1-5].Clinical features of the disorder
include movement disorders, neurological symptoms, cutaneous and conjunctival telangectasias,
possible increased risk of malignancy, and immunodeficiency [2,3].
Due to the involvement of the ATM gene in cell cycle progression and DNA repair, A-T
patients are sensitive to ionizing radiation and have an increased susceptibility to cancer,
particularly to hematolymphoid malignancies [1-10]. Immunodeficiency in A-T often involves
T-cell lymphopenias, thymic hypoplasia, and deficiencies in immunoglobulin production, mainly
IgA, IgE, and IgG2, which places A-T patients at an increased risk for recurrent sinopulmonary
infections [1-3,10].
An additional consequence of mutations in the ATM gene is a predisposition to gammopathies,
both monoclonal and polyclonal [1,10-13]. In a recent study, 39% of A-T patients showed
hypergammaglobulinemia, with 8% of patients having a monoclonal or oligoclonal gammopathy
[1,12]. The differential diagnosis for monoclonal gammopathies in the general pediatric population
includes congenital, autoimmune, and infectious diseases, hematologic conditions, solid organ
malignancies, and renal or hepatic disease [12]. However, no studies have specifically described
the differential diagnosis for gammopathies in A-T patients. Here in, we describe a case of a child
with A-T who was found to have a monoclonal gammopathy and propose a preliminary differential
diagnosis for monoclonal gammopathy in A-T patients.
Case Report
The patient is a pediatric patient with a past medical history of A-T, IgA deficiency, presumed
epilepsy, and asthma who was born at term to a 26 year old G1P1 mother. The patient's family
history was contributory for A-T in a sibling and a treatment with leg braces for undocumented conditions in distant relatives. The patient experienced irregular
breathing postnatally and was observed in the neonatal intensive
care until discharge at 5 days old. The patient met developmental
milestones within the first few months of life. However, at 1 year
of age, the child’s parents noted that the patient began walking but
preferred to toe-walk with knees hyperextended, resulting in falls.
This prompted a neurological evaluation. Brain magnetic resonance
imaging, cerebrospinal fluid studies, complete metabolic panel,
creatine phosphokinase, lactic acid, lysosomal enzyme battery, very
long chain fatty acid levels, and an acyclcarnitine profile were all
normal. However, IgA was found to be absent (normal 15 mg/dl to
241 mg/dL) and alpha-fetoprotein was found to be elevated at 87 ng/
mL (normal < 20 ng/mL). The patient was referred to genetics for
sequencing of the ataxia-telangiectasia mutated gene.
Full gene sequencing of the patient's ATM gene was performed
and two alterations were detected: a variant heterozygous change
from G to A at nucleotide 331+1 of the ATM gene (c.331+1 G>A)
and a positive heterozygous change from C to A at 1931, resulting in
a nonsense change at codon 644 (c.1931 C>A; p.Ser644*). The first
alteration involved the highly conserved canonical splice donor site of
intron 6 (also known as intron 4). In silico splicing analyses [14] predict
that this alteration would obliterate the normal splice donor site and
a different pathogenic ATM mutation was previously documented at
this site [15]. The second alteration results in premature termination
of the transcript, which is an alternation previously documented in
A-T [16]. Both mutations were predicted to be deleterious [14-16].
At diagnosis, total serum IgG was elevated at 1340 mg/dL
(580 mg/dL to1256 mg/dL), with IgG1-IgG4 subtypes within
normal limits. Total serum protein was elevated at 8.6 g/dL (6.0 g/
dL to8.0 g/dL). The patient also had a neutrophilia of 81% (12%
to 34%) and lymphopenia of 8% (45% to 75%). Serum Protein
Electrophoresis (SPEP) was performed which showed a monoclonal
protein (M-protein, M-spike, monoclonal gammaglobulin)
detected with an approximate concentration of 0.56 g/dL (Figure
1). Immunoelectrophoresis (IEP) identified the monoclonal protein
as either IgG λ or free λ (Figure 2). Repeat SPEP and IEP showed
a monoclonal protein at an approximate concentration of 0.32 g/dL
that was either IgG λ or free λ light chain. Serum IEP for IgD and IgE
was also performed, which showed no IgD λ or IgE λ.
Three sets of serum free light chain testing were performed and
showed a mean κ concentration of 16.0 mg/L ± 1.7 mg/L (Mean ±
SEM) (normal 3.3 md/L to 19.4 mg/L) and a mean λ concentration of
10.3 mg/L ± 0.3 mg/L (normal 5.7-26.3 mg/L). The average κ:λ ratio
was 1.54 ± 0.1 (normal range 0.26-1.65).
The patient was referred to hematology-oncology for further
testing, but it was felt that the patient’s risk of malignancy was low
due to a normal lymph node exam, and complete blood count, lactate
dehydrogenase level, and uric acid level that were within normal
limits. The patient will, however, continue to be closely monitored
henceforth for changes in clinical status.
Figure 1
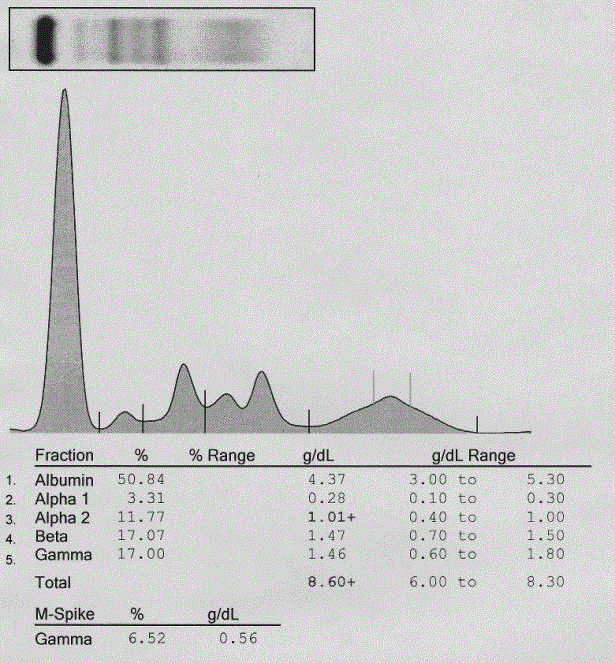
Figure 1
Monoclonal spike on serum protein electrophoresis.
Protein electrophoresis depicting relative and absolute concentrations of
serum proteins after densitometric evaluation of SPEP areas. A blood sample
was drawn from the patient and analyzed for total serum proteins on SPIFE
3000 (Helena, Beaumont, TX, USA)High Resolution Protein Electrophoresis
equipment per the manufacturer’s protocol. A monoclonal spike is present
in the gamma region at a concentration of 0.56 g/dL. Mild hemolysis of the
sample is indicated by the peak between the alpha 2 and beta regions.
Figure 2
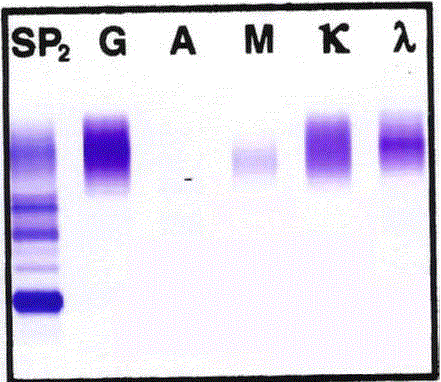
Figure 2
Monoclonal lambda light chain band on serum protein
immunoelectrophoresis.
Immunoelectrophoresis (serum protein electrophoresis with immunofixation)
depicting relative concentrations and clonality of serum proteins. A blood
sample was drawn from the patient, serum was isolated and serum protein
electrophoresis with immunofixation was performed on SPIFE 3000 (Helena,
Beaumont, TX, USA) High Resolution Protein Electrophoresis equipment per
the manufacturer’s protocol. A distinct band is evident for the lambda light
chain amid a polyclonal background. It is possible that a distinct IgG band
is present but it is hidden by the predominantly polyclonal IgG staining. IgM
is polyclonal, as is the kappa light chain. The barely visible polyclonal IgA
population is consistent with the patients known IgA immunodeficiency.
Discussion
A-T is an autosomal recessive disorder arising from mutations in
the ATM gene that result in neurodegeneration, cutaneous and ocular
telangectasias, cancer susceptibility, and immunodeficiency [2-3].
Clinically, the neurodegeneration manifests as oculomotor apraxia,
dysarthria, and movement disorders such as choreo-athetosis,
dystonia, Parkinsonism, among other neurological dysfunctions [2-
3].
Due to the involvement of the ATM gene in cell cycle progression
and DNA repair, homozygous mutated A-T patients are sensitive to
ionizing radiation and may have an increased risk of hematologic
or gastric malignancy, dysgerminoma, medulloblastoma, and
gonadoblastoma, among other cancers [1-9]. A-T patients are most
likely to develop hematologic malignancies, with Caucasian A-T
patients and African-American A-T patients carrying a 250-fold and
750-fold increased risk of lymphoma, respectively, as compared with
the general population [5,10]. There is an increased risk of developing
both T and B cell tumors, with B cell non-Hodgkin’s lymphoma being the most common B cell tumor and T acute lymphocytic leukemia,
T cell lymphoma, and T prolymphocytic leukemia, being the most
common T cell neoplasms [5]. Additionally, female carriers of an
ATM gene mutation have a documented increased risk of breast
cancer [3,17].
Immune deficiency in A-T is variable in each individual patient
but also in one patient across time. The most common immune
defects in A-T involve cellular and humoral immunity: CD4+T
cell lymphopenia, reduced delayed-type hypersensitivity reactions,
and deficiencies in IgA, IgE, and IgG2 [1,10]. Thymic hypoplasia
is observed as an absence of Hassall’s corpuscles and decreased
corticomedullary differentiation [1]. Lymphocytes of A-T patients
exhibit telemeric erosion and fusions, as well as cell cycle dysfunction,
which may also play a role in immunodeficiency in A-T [18]. Due to
these factors, A-T patients have a predilection for recurrent bacterial
sinopulmonary infection which, worsened by neurodegenerative
dysphasia, leads to the most common cause of death in the disorder:
aspiration pneumonia [1-3].
Additional sequelae of ATM gene mutations are monoclonal
and polyclonal gammopathies [1,10-13]. Gerritsen et al. [11] studied
monoclonal gammopathies in the general pediatric population. They
detected all immunoglobulin isotype monoclonal gammopathies
except for IgA monoclonal gammopathies and identified a
predominance of lambda light chain gammopathies. Conversely
Akha et al. specifically studied gammopathies in A-T patients. They
found that 39% of A-T patients showed hypergammaglobulinemia,
with 8% of patients having a monoclonal gammopathy [1,12]. They
also found that all immunoglobulin isotypes were represented in A-T
patients with monoclonal gammopathy and did detect A-T patients
with lambda light chain gammopathies, although the lambda light
chain did not predominate [1,12].
Our patient exhibited a monoclonal gammopathy involving the
lambda light chain, but the immunoglobulin isotype was unable to
be determined. It is unlikely that the gammopathy represented free
light chain lambda, as the free κ:λ ratio was only mildly elevated in
two measurements, with the mean ratio being within normal limits.
Urine protein electrophoresis and immunoelectrophoresis would be
helpful to further characterize the isotype, however the clinical team
did not order these studies. Although our data cannot be directly
compared to the Gerritsen et al. study, as they were not specifically
evaluating A-T patients, our data is in agreement with Akha et al.’s
finding that A-T patients can exhibit lambda light chain monoclonal
gammopathies.
The differential for monoclonal gammopathies in the general
pediatric population has been established by Gerritsen et al. and
Karafin et al. and includes congenital, autoimmune, and infectious
diseases, hematologic conditions, solid organ malignancies, and
renal and hepatic diseases [11,12]. In this classification, A-T is
included in the spectrum of congenital diseases. It is likely that the
causes of gammopathies in A-T patients may predominantly include
malignancy, autoimmunity, and infection, considering the unique
susceptibility of these patients to cancer and immunodeficiency.
Indeed, case reports published on gammopathies in A-T patients
have described prior oral and genital herpetic infections and diffuse
plasmocytosis of the kidney, liver, bone marrow, and lungs [10,13].
It has been hypothesized that monoclonal and polyclonal
gammopathies in A-T may result from exposure of lymphocytes to
events that increased double stranded DNA breaks, such as ionizing
radiation, chemotherapy, or infections [1,11]. The lack of repair
of these breaks, coupled with abnormal thymic development and
defective cell cycle regulation, could then lead to clonal dysregulation
of B cells and gammopathies [1]. Data supporting this includes
abnormalities in TCR rearrangements in A-T and an increased
incidence of translocations involving TCR and immunoglobulin
genes [3,8]. In fact, these translocations can be detected in 10% of
circulating T cells in A-T patients throughout their lifetime [19]. In
many cases, these monoclonal gammopathies appear to be short-lived
[11], as is the case in the general pediatric population [11]. However,
it is wise for clinicians to be aware of the unique susceptibility of A-T
patients to malignancy and immunodeficiency and screen for the
possibility of an underlying malignancy, autoimmune disorder, or
infection.
References
- Akha AS, Humphrey RL, Winkelstein JA, Loeb DM, Lederman HM. Oligo-monoclonal gammopathy and hypergammaglobulinemia in ataxia-telangiectasia. Medicine. 1999;78(6):370-81.
- Levy A, Lang AE. Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov Disord. 2018.
- Mavrou A, Tsangaris GT, Roma E, Kolialexi A. The ATM Gene and Ataxia Telangiectasia. Anticancer Research. 2008; 28(1B): 401-5.
- Ball LG, Xiao W. Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacol Sin. 2005;26(8):897-907.
- Boultwood J. Ataxia telangiectasia gene mutations in leukemia and lymphoma. J Clin Pathol. 2001;54(7):512-6.
- Hall J. The Ataxia-telangiectasia mutated gene and breast cancer: Gene expression profiles and sequence variants. Cancer Lett. 2005;227(2):105-14.
- Wang L, Wang Q-T, Liu Y-P, Dong Q-Q, Hue H-J, Miao Z, et al. ATM signaling pathway is implicated in the SMYD3-mediated proliferation and migration of gastric cancer cells. J Gastric Cancer. 2017;17(4):295-305.
- Lipkowitz S, Stern MH, Kirsch, IR. Hybrid T cell receptor genes formed by interlocus recombination in normal and ataxia-telangiectasia lymphocytes. J Exp Med. 1990;172:409-18.
- Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, et al. Immunoglobulin class switch recombination is impaired in atm-deficient mice. J Exp Med. 2004;200(9):1111-21.
- McDonald PS, Cora-Bramble D, De Palma L. Monoclonal gammopathy of the immunoglobulin A class in a two-year-old with ataxia telangiectasia. Pediatr Develop Pathol. 1998; 1:319-21.
- Gerritsen E, Vossen J, van Tol M, Jol-van der Zijde C, Van der Weijden-Ragas R, Radl J. Monoclonal gammopathies in children. J Clin Immunol. 1989;9(4):296-305.
- Karafin MS,Humphrey RL, Detrick B. Evaluation of monoclonal and oligoclonal gammopathies in a pediatric population in a major urban center. Am J Clin Pathol. 2014;141(4):482-7.
- Cawley LP, Schenhen JR. Monoclonal hypergammaglobulinemia of the gamma-M type in a 9-year-old girl with ataxia-telangiectasia. Am J Clin Pathol. 1970;54(6):790-801.
- ClinVar. NM_000051.3(ATM):c.331+1G>A AND Ataxia-telangiectasia syndrome. National Center for Biotechnology Information. 2016 .
- Cavalieri S, Funaro A, Pappi P, Migone N, Gatti RA, Brusco A. Large genomic mutations within the atm gene detected by mlpa, including a duplication of 41 kb from exon 4 to 20. Ann Hum Genet. 2008:72(pt 1);10-8.
- Li A, Swift M. Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. 2000;92(3):170-7.
- Geoffroy-Perez B, Janin N, Osian K, Lauge A, Croquette MF, Griscelli C, et al. Cancer risk in heterozygotes for Ataxia-Telangiectasia. Int J Cancer. 2001;93(2):288-93.
- Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd PJ, et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13(3):350-3.
- Lavin MF, Gueven N, Bottle S, Gatti RA. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. British Medical Bulletin. 2007; 81-82 (1): 129-47.