Case Report
Von Recklinghausen’s Disease with a Typical Features
Juan Antonio Garcia-Rodriguez*
Department of Family Medicine, University of Calgary, Canada
*Corresponding author: Juan Antonio Garcia-Rodriguez, Department of Family Medicine, Cumming School of Medicine, University of Calgary, Sunridge Family Medicine Teaching Centre 2685 – 36 Street NE, Calgary, Alberta, Canada
Published: 08 Aug, 2017
Cite this article as: Garcia-Rodriguez JA. Von
Recklinghausen’s Disease with a
Typical Features. Ann Clin Case Rep.
2017; 2: 1414.
Abstract
A 42-year-old man consulted complaining of an irritated skin-colored nodule to the left side of his head that was identified as a neurofibroma. Numerous similar lesions were found to his torso and neck. He had no dysmorphic features or bone abnormalities, but he was found to have left axilla and right inguinal freckling, long soft-brown asymmetric skin patches of irregular borders on his torso that crossed the midline with no fullness under the skin. His left iris had some orange-brown colored specks. He had normal blood pressure and no neurological abnormalities. MRI of his body showed no plexiform neurofibromas. Nobody else in his family had similar findings. Based on his clinical findings, a diagnosis of a mosaic form of Neurofibromatosis type 1 (von Recklinghausen’s disease) was made, as a de novo mutation.
Case Presentation
A 42-year-old man consulted complaining of a longstanding soft flesh-colored nodule on the left side of his head that was irritated and sometimes bled after combing his hair (Figure 1). He remembered having a similar lesion removed from his chest area 15 years prior in another country that was not sent for pathologic study. His physical examination revealed numerous similar skincolored nodules on his neck, behind both ears and torso (Figures 2 and 3). His exam demonstrated neither dysmorphic features, nor scoliosis or tibial bowing. Freckling was found to his left axilla (Figure 4) and right inguinal areas, as well as larger soft-brown confluent patches of skin with irregular borders covering the majority of his shoulders and torso and crossing the midline (Figures 2 and 3). No fullness was palpated under the skin covered by the maculas. Some orange-brown colored specks were appreciated in the bottom half of his left iris (Figure 5); none were present on his right iris. Blood pressure was normal and no neurological abnormalities (hearing or visual changes, ataxia, weakness, swallowing difficulties or changes in sensation) were present. He suffered from occasional headaches. Neither his parents nor siblings had similar findings. The findings on physical examination suggested a diagnosis of neurofibromatosis type 1 (von Recklinghausen’s disease). Three of his nodular skin lesion were biopsied and sent to pathology; the pathology report confirmed the diagnosis of neurofibromas with no malignant changes. The patient underwent MRI assessment of his chest, abdomen, pelvis and thorax encountering no plexiform neurofibromas. His clinical features and unusual path of skin findings suggested a mosaic form of the disease and the lack of family history suggested a de novo mutation. The patient underwent clinical genetic evaluation and no molecular testing was deemed necessary due to the technical difficulties inherent to his mosaic pattern, and as it would not change his diagnosis or management. His three children were examined and none of them had any features of neurofibromatosis, his younger child was 4 years old with no evident skin changes (clinical findings are expected to be found by the age of 3 or 4). The patient continues in a longstanding monitoring, in particular for further skin changes, neurological symptoms and for the possibility of developing a pheochromocytoma.
Discussion
Neurofibromatosis affects 1 in 3500 people, is a multisystem
autosomal dominant condition where tumors are developed along
the course of peripheral nerves, and soft-tissue and bone deformities
can also be present [1,2]. The majority of patients are asymptomatic
but some can present with neurological or bone complains. The
expression of the disease is highly variable among family members
with the same mutation [3]. The diagnosis is made on a clinical basis
although mutational analysis and molecular testing are now available
for specific cases [1].
The described nodular lesions are neurofibromas, which are
benign tumors of Schwann cells, perineural cells and fibroblasts. The
superficial cutaneous manifestations are hallmarks of this disease.
In some cases, firm subcutaneous neurofibromas can be found.
Diffuse subcutaneous masses (diffuse plexiform neurofibromas) can
produce disfiguring deformities, and lesions involving spinal nerve
roots (nodular plexiform neurofibromas) can produce neurological
symptoms. Neurofibromas have the potential to become malignant.
The patients suffering from this disease also present with skin
maculae ranging from soft-brown (café-au-lait spots) to freckling
patches that can be appreciated in the trunk, pelvis or extremities [4].
Skin fold freckling (Crowe sign) is the most specific finding for this
condition [1]. The mentioned eye findings (orange-brown colored
specks) are Lisch nodules which are benign hamartomas that can be
seen without magnification. Slit lamp examination can differentiate
them from nevi on the iris by demonstrating elevated lesion instead of
flat ones. Other associated ophthalmologic findings are optic gliomas
[2]. Lisch nodules occur in 90% of adults with neurofibromatosis 1.
Optic gliomas that can alter color vision and can produce progressive
sight loss [1]. Skin findings are usually bilateral but asymmetrical
presentation can be seen in mosaic forms of the disease (when not all
of the patient’s cells carry the genetic change in the NF-1 gene) as is
the case of the presented patient. In 50% of cases the genetic mutation
is inherited from a parent and 50% of the time the mutation is de
novo [6,7].
Cognitive problems can be present in 60% of cases including
difficulties at school, learning disabilities or attention deficits.
Possible associated neurological symptoms include weakness,
numbness and paresthesias in any extremities or parts of the body,
as well as headaches in 20% of patients and seizures in up to 10%
[2]. Some cardiovascular manifestations include hypertension, which
suggests renal artery stenosis (1% of patients), and vascular dysplasias
most frequently affecting the aorta and carotid arteries [1]. Bone
abnormalities include scoliosis in 10% of patients [5], pseudoarthrosis
(mainly of the lower third of tibia and fibula), bowing of legs, sphenoid
wing dysplasia, fibrous dysplasia and subperiosteal bone cysts [1,4].
If the patient presents with neurological symptoms, an MRI or CT
scan of the affected area can determine the presence of underlying
plexiform neurofibromas. Treatment is required if the neurofibromas cause severe symptoms, disfiguration or if they are irritated by their external location. Surveillance for malignancy is recommended.
Figure 1
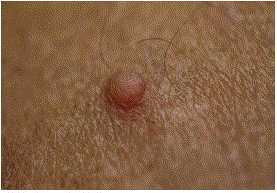
Figure 1
Nodular skin lesion – Neurofibroma.
Figure 2
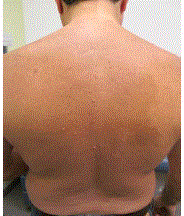
Figure 2
Skin lesions – back.
Figure 3
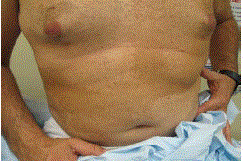
Figure 3
Skin lesions – front.
Figure 4
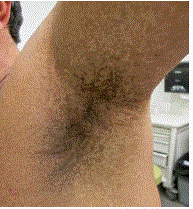
Figure 4
Skin fold freckling (Crowe sign).
Figure 5
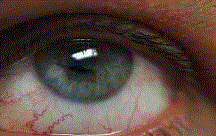
Figure 5
Eye findings - Lisch nodules.
Table 1
Table 1
Differential Diagnosis: Diagnostic Features of Neurofibromatosis type 1 and 2*.
Authors’ Contributions
JAGR is the primary health provider for the patient, conceived, designed, compiled the data for the article and wrote the article.
Authors’ Information
JAGR is the primary health provider for the patient, who practices Family Medicine and Sports Medicine. He is an Assistant Professor at the Family Medicine Department of the University of Calgary.
References
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009; 61: 1-14.
- Tonsgard JH. Clinical manifestations and management of neurofibromatosis type 1. Semin Pediatr Neurol. 2006; 13: 2-7.
- Korf BR, Rubenstein AE. Neurofibromatosis: A handbook for patients, families, and health care professionals. 2nd ed. New York: Thieme; 2005.
- Riccardi VM. Neurofibromatosis: phenotype, natural history and pathogenesis. John Hopkins University Press, Baltimore and London; 1992.
- Crawford AH. Pitfalls of spinal deformities associated with neurofibromatosis in children. Clin Orthop Relat Res. 1989; 245: 29-42.
- Theos A, Korf BR; American College of Physicians; American Physiological Society. Pathophysiology of neurofibromatosis type 1. Ann Intern Med. 2006; 144: 842-849.
- Hoa M, Slattery WH. Neurofibromatosis 2. Otolaryngol Clin North Am. 2012; 45: 315-332.