Case Report
Atypical Alagille Syndrome Diagnosed by Genomic Analysis
Zhen Zhen Zhang1*, Kun Zhu2*, QuanBo Liu1 and HongMei Xu1*#
11Department of Infectious Disease, Children’s Hospital of Chongqing Medical University, China
2Department of Radiology, Children’s Hospital of Chongqing Medical University, China
#Authors contributed equally to this paper
*Corresponding author: Hongmei XU, Department of Infectious Disease, Children’s Hospital of Chongqing Medical University, No.136, Zhong Shan Er Road, Yu Zhong District, Chong Qing, 400014, China
Published: 14 Jun, 2017
Cite this article as: Zhang ZZ, Zhu K, Liu Q, Xu H. Atypical
Alagille Syndrome Diagnosed by
Genomic Analysis. Ann Clin Case Rep.
2017; 2: 1377.
Abstract
Alagille Syndrome (ALGS) is an autosomal dominant disease presented by multi-system disorder including liver, heart, eyes, vertebrae, and face. Traditionally, the diagnosis of ALGS is based on a combination of liver biopsy with at least three of the major clinical features. However, this may cause misdiagnosis of atypical or mild ALGS. Here we reported a 12-year-old boy with chief complaint about recurrent mild transaminase elevation and hepatosplenomegaly over 11 years. There were no jaundice and other system disorders. He also had liver biopsy which didn’t present paucity of intrahepatic bile duct. The genome testing demonstrated a heterozygous mutation on exon12 of JAG1 gene. The boy was finally diagnosed as ALGS and treated by ursodeoxycholic acid as well as fat-soluble vitamins. For patients with chronic and progressive liver damage, ALGS should be taken into consideration. Molecular diagnosis is particularly useful for patients with atypical or mild Alagille syndrome who do not meet classic diagnostic criteria.
Abbreviations
ALGS: Alagille Syndrome; RL: Right Lobe; LL: Left Lobe; CL: Caudate Lobe; G: Gallbladder; PV: Portal Vein; TB: Total Bilirubin; DB: Direct Bilirubin; ALB: Albumin; ALT: Alanine Aminotran; AST: Aspartate Aminotransferase; AKP: Alkaline Phosphatase; GGT: γ-GlutamylTranspeptidase; LDH: Lactate Dehydrogenase; BA: Bile Acid
Introduction
Alagille Syndrome (ALGS) is an autosomal dominant disease presented by multi-system disorders including liver, heart, eyes, vertebrae, and face [1]. ALGS is caused by mutations in or deletions of either the JAGGED1 (JAG1) or NOTCH2 gene [2,3]. Traditionally, the diagnosis of ALGS is based on a combination of the presence of intrahepatic bile duct paucity on liver biopsy with at least three of the major clinical features including liver disease (cholestasis), congenital heart defects, vertebral abnormalities, ocular anomalies and characteristic facial features [4]. However, this may cause misdiagnose with atypical or mild ALGS. Here we reported a 12-year-old male patient presented by recurrent transaminase elevation and hepatosplenomegaly over 11 years. There were no jaundice and other system disorders. Liver biopsy showed the patient had liver fibrosis but not the paucity of intrahepatic bile duct. The genome sequencing demonstrated a heterozygous mutation of JAG1 gene. The patient was finally diagnosed as ALGS. The availability of molecular testing led to the diagnosis of ALGS precisely.
Patient Information
A 12-year-old male patient was charged into hospital with chief complaint about recurrent
transaminase elevation and hepatosplenomegaly over 11 years, which was the third time of his
hospitalization. 11 years ago, the patient was found anorexia, abdominal distention, and mass
on the left upper abdominal. There weren’t obvious jaundice, fever, vomits and clay stool. The
patient’s liver function showed a mild elevation of transaminase while the CT scanning showed
hepatosplenomegaly accompanied with loss of normal ratio of hepatic lobes. The patient also
received gastroscopy examination which indicated mild esophageal varix. Finally, the patient was
diagnosed with liver cirrhosis. His parents rejected further treatment and only treated with Chinese
herbal medicine (no details of the herbal medicine). 5 years ago, the patient admitted to hospital
again with poor appetite and pale complexion. Likewise, the patient didn’t have other symptoms.
The liver function still presented with a mild transaminase elevation.
The patient received liver biopsy this time which showed swelling,
ballooning degeneration of hepatocyte, focal necrosis and lymphocyte
infiltration around portal tract. Neither fibrous hyperplasia nor bile
ducts paucity were found. The patient was treated with injected
reduced glutathione for a week and then changed into oral formula.
The patient’s liver function fluctuated continuously afterward and
hospitalized for the third time.
The patient was G2P2 and the birth weight is 3.8 Kg. The patient
had delayed clearance of jaundice which lasted nearly 6 months after
birth. There was neither growth nor mental retardation of the patient.
The patient had an elder brother who was health.
Figure 1
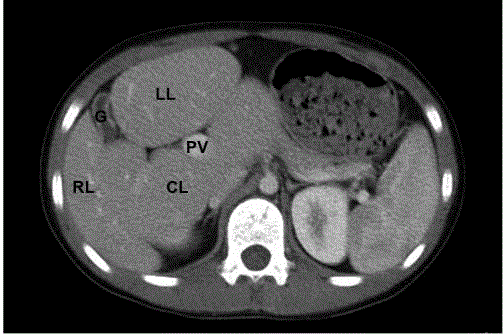
Figure 1
abdomen CT scan of the patient. There was loss of
normal ratio among liver lobes, with lessen of right lobe and enlargement of
left and caudate lobe. The portal vein was on the upper limit.
Abbreviations: RL: Right Lobe; LL: Left Lobe; CL: Caudate Lobe; G:
Gallbladder
Figure 2
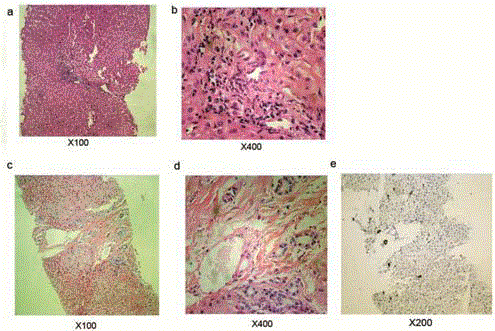
Figure 2
Liver histology results of the patient. a,b: liver histology by
hematoxylin and eosin staining 5 years ago; c,d: latest liver histology by
hematoxylin and eosin staining; e: Ki67 staining of bile duct.
Table 1
Table 1
laboratory parameters of patient’s liver function on different time point.
Clinical Findings
The patient had peculiar facial features with prominent forehead, bulbous nose, and pointed chin (supplementary data 1). There were no jaundice, pedal edema, purpura, spider nevi, cardiac murmur and lung rale. Abdomen examination revealed enlargement of liver and spleen. The liver was firm in texture and the span was 4.5 cm along the right mid-clavicular line while the spleen was 4 cm below the left costal margin with firm texture.
Diagnostic Assessment
There was slight elevation of liver transaminase, other blood tests including Alpha-Fetoprotein (AFP), ceruloplasmin, cholesterol, and triglyceride level, coagulation function test, blood ammonia, Tandem mass spectrometry were normal. The HBsAg, anti-HCV, and autoantibody were all negative. CT scan indicated loss of normal ratio among liver lobes, with lessening of right lobe and enlargement of left and caudate lobe (Figure 1). The portal vein was on the upper limit. Other Imaging examinations including chest X-ray and cardiac ultrasonic were normal. Liver biopsy demonstrated less swelling, ballooning degeneration, focal necrosis of liver cells, however, there was mild proliferation of collagen and bile canaliculus around portal areas. The gene analysis found the boy had a heterozygous mutation of JAG1 gene on exon12 with the deletion of cytosine on p1563-1564, which caused frame shift mutation.
Therapeutic Intervention
The patients were diagnosed with Alagille Syndrome (ALGS) and treated with ursodeoxycholic acid and fat soluble vitamins. The patient was asked to follow-up regularly.
Discussion
In this case, hepatic disorder (Table 1) and facial feature were two main manifestations of the patient. No positive found on other systems. There wasn’t obvious jaundice except for prolonged neonatal jaundice. Liver biopsies didn’t show paucity of bile ducts, there was mild proliferation of bile canaliculus around portal areas instead (Figure 2). This result indicated that the liver histology of ALGS could be varied. Some patients may not present as paucity of bile duct in histology. In fact, it has been realized that a liver biopsy is no longer considered mandatory to make a diagnosis of ALGS, and the presence of cholestasis is acceptable to fulfill this criterion. The γ-Glutamyl transpeptidase and bile acid level of the patient were kept abnormal, which demonstrated the patients had chronic cholestasis. Except for that, the latest liver biopsy showed fibroplasias, which was not obvious before. This revealed that the liver damage was continuously progressing even if the symptom and liver function not further aggravated. We excluded other chronic liver diseases including Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD), Glycogen storage disease, Wilson's disease, Progressive Familial Intrahepatic Cholestasis III (PFIC-III) and Autoimmune liver disease through blood test, imaging examination, and gene test. We also did the gene test of the patient’s parents since the patient is diagnosed. The result proved that his parents didn’t carry the same mutation. The patient had a de novo mutation. Supportive treatment is the main therapy for live damage caused by alagille syndrome. Ursodeoxycholic acid is recommended for cholestasis. It has been reported that liver transplantation for ALGS has an 80% 5-year survival rate, and results in some catch-up growth in 90% of affected individuals [5].
Conclusions
Alagille syndrome has complicated clinical manifestations varies from sub-clinical to multisystem disorders. For patients with chronic and progressive liver damage in his early life, ALGS should be taken into consideration. Molecular diagnosis is particularly useful for patients with atypical or mild Alagille syndrome who do not meet classic diagnostic criteria.
Informed Consent
Written informed consent was obtained from the patient’s legal guardians for publication of this case report and any accompanying images.
References
- Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia with characteristic facies, vertebral malformations, retarded physical, mental and sexual development and cardiac murmur. J Pediatr. 1975; 86: 63-71.
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016: 975-982.
- Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez-Lara PA, Piccoli DA, et al. Jagged1 (JAG1) mutations in Alagille syndrome: Increasing the mutation detection rate. Hum Mutat. 2006: 27: 436–443.
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006; 79: 169-173.
- Sheflin-Findling S, Arnon R, Lee S, Chu J, Henderling F, Kerkar N, et al. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012; 47: 1453-1456.