Case Report
Yamaguchi’s Disease in African American Patient: A Case Report and Literature Review
Dawit Kibru Worku1* and Michael Gebre Yohannes Tedla2
1Bahir Dar University, Ethiopia
2Second Year Internal Medicine Resident, Mountain View Hospital, USA
*Corresponding author: Dawit Kibru Worku, Bahir Dar University, PO Box 1345, Bahir Dar, Amhara, Ethiopia
Published: 07 Jun, 2017
Cite this article as: Worku DK, Tedla MGY. Yamaguchi’s
Disease in African American Patient:
A Case Report and Literature Review.
Ann Clin Case Rep. 2017; 2: 1370.
Abstract
Introduction: Apical hypertrophic cardiomyopathy is a rare form of hypertrophic cardiomyopathy which usually involves the apex of the left ventricle and rarely involves the right ventricular apex or both. Apical hypertrophic cardiomyopathy is very rarely treated surgically. This made our patient
among the few who underwent surgical intervention due to outflow obstruction.
Case Presentation: A 45 years old African American female patient with past medical history of
Hypothyroidism who presented at Mountain View Hospital with recurrent episodes of presyncope
and syncope, palpitation, dyspnea and mild precordial chest pain. On physical exam, vital signs
were normal, cardiac exam showed no sign of murmur or any sign of cardiac decompensation.
Patient was thoroughly worked up.
Conclusion: Counseling and management of patients with apical hypertrophic cardiomyopathy
should be planned and followed.
Keywords: Apical hypertrophic cardiomyopathy; Cardiology; Case report; Hypertrophic cardiomyopathy
Abbreviations
AHCM: Apical Hypertrophic Cardiomyopathy; CCT: Cardiac Computed Tomography; CMR: Cardiovascular Resonance Imaging; EKG: Electrocardiogram; HCM: Hypertrophic Cardiomyopathy; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; SPECT: Single Photon Emission Computed Tomography; TTE: Trans-Thoracic Echocardiogram
Introduction
Apical Hypertrophic Cardiomyopathy (AHCM) is a rare form of Hypertrophic Cardiomyopathy
(HCM) which usually involves the apex of the left ventricle and rarely involves the right ventricular
apex or both [1]. In AHCM detectable sarcomere protein gene mutations could be less prevalent in comparison with other forms of HCM [2]. These patients do not have left ventricular outflow obstruction but may have mid-ventricular obstruction [3,4]. Mid-ventricular obstruction is
extremely rare in AHCM. We believe reporting of such obstruction may give insight in the prognosis
of AHCM, and clinicians diagnosing and treating ACHM patients should be alerted.
Morphologically AHCM is divided into 3 types: pure focal, pure diffuse and mixed, of which
pure focal is most common. In a Japanese cohort, patients with a pure focal phenotype had improved
survival over a pure diffuse phenotype. This morphologic sub-classification was recently recognized
in North America [5]. However in clinical practice this sub-classification is not widely accepted and its clinical relevance is unknown. Others have divided AHCM into two groups, based on whether
they had isolated asymmetric apical hypertrophy (pure AHCM) or had co-existent hypertrophy
of the interventricular septum (mixed AHCM) [6]. The pure AHCM is predominant in Japanese
patients, while the mixed form has been linked to Caucasian patients [2].
Sudden cardiac death is rare in patients with isolated AHCM, and overall cardiovascular
morbidity may be less common compared with other HCM phenotypes. Nonetheless, available
outcomes studies are insufficiently powered for robust conclusions and certain AHCM patients are
at high risk of cardiovascular morbidity [6].
Case Presentation
A 45 years old African American female patient with past medical history of Hypothyroidism who presented at our Hospital with recurrent episodes of presyncope and syncope, palpitation, dyspnea and mild precordial chest pain. On physical exam, vital signs were normal, cardiac exam showed no sign of murmur or any sign of cardiac decompensation. Patient was thoroughly worked up with: EKG showed Left Ventricular (LV) hypertrophy and T wave inversion on precordial leads (Figure 1). Chest X-ray and Cardiac enzymes were normal. Trans-thoracic Echocardiography showed Left Ventricular Ejection Fraction (LVEF) of 55% with LV apical hypertrophy Perfusion study showed LVEF of 54%, good perfusion, no ischemic EKG changes. Cardiac MRI showed thickened LV myocardium involving most of the LV, only sparing a small portion of the bases (Figure 2). Cardiac catheterization showed no significant coronary artery disease, 11mmHg mid cavity gradient; unable get into apex due to cavity obliteration (Figure 3a and b). With the diagnosis of AHCM, She was started on Beta Blocker for 3 months but she developed severe bradycardia. She was then referred to Mayo clinic at Arizona where she underwent septal myomectomy.
Figure 1
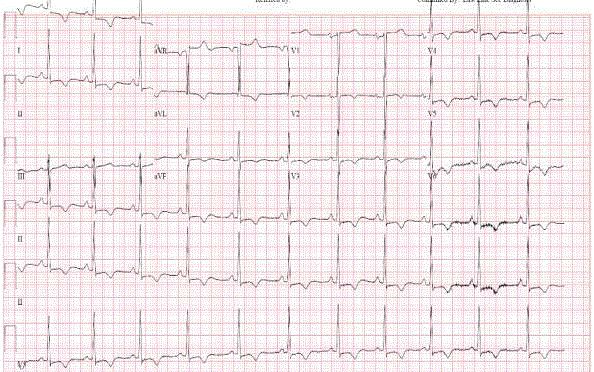
Figure 1
EKG of our patient showing diffuse precordial T wave inversion
and LV hypertrophy.
Figure 2
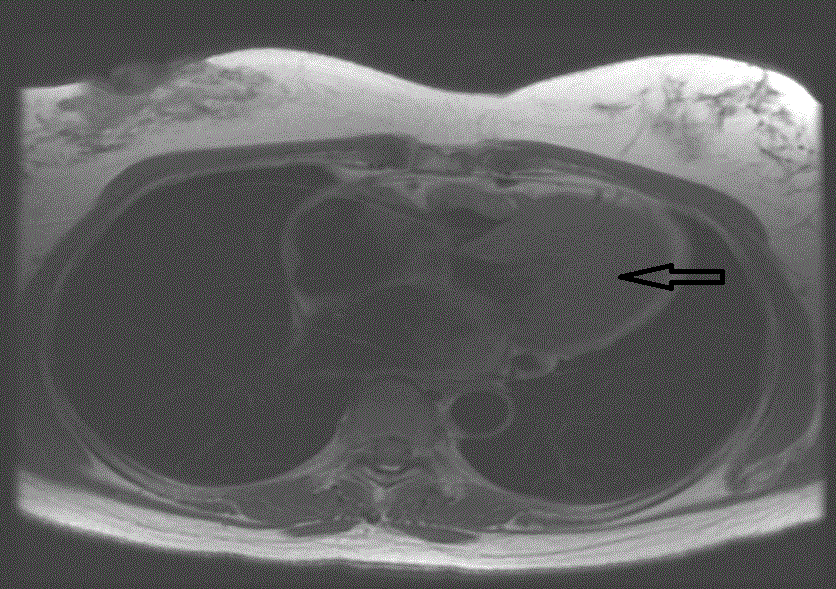
Figure 2
Cardiac MRI showing significant Left ventricular apical wall
hypertrophy (arrow).
Figure 3
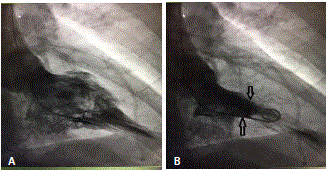
Figure 3
Left ventriculography showing LV filling during a) Diastoly and b)
Systoly.
Discussion
Epidemiology
Ethnically, AHCM accounts for 13-41% of all variants of HCM
among Asian individuals, whereas the prevalence among non-Asians
is < 5%. Of all the HCM patients in Japan the prevalence of AHCM
was 15%, whereas in USA the prevalence was only 3% [7] However,
much of these epidemiologic data are derived from studies conducted
in the early 1990s, and the exact prevalence of AHCM in non-Asians
may be underestimated due to diagnostic unawareness as well as the heterogeneous appearance on trans-thoracic echocardiogram, the most frequently utilized diagnostic modality [8].
Diagnosis
Most patients with AHCM are diagnosed incidentally or with
mild symptoms including fatigue, fainting, chest pain and palpitation,
although cases were reported with symptoms of angina, myocardial
infarction, heart failure, atrial or ventricular fibrillation. In patients
presenting with severe symptoms, apical myomectomy can be done
[3,4].
The diagnostic criteria for AHCM include demonstration of
asymmetric left ventricular hypertrophy confined predominantly
to the LV apex. This hypertrophy must present with an apical wall
thickness >=15mm and a ratio of maximal apical to posterior wall
thickness ≥1.3. This can be based on either echocardiogram with or
without contrast medium. Such medium improve endocardial border
visualization or cardiovascular resonance imaging (CMR) [2].
The preferred initial imaging test is a trans-thoracic
echocardiogram (TTE). Image quality can vary however, as it is
dependent on many factors including body habitus and the skill of the
sonographer to focus on the left ventricular apex. When the baseline
images are suboptimal, a contrast echocardiogram may be utilized
depicting a “spade-like” configuration of the apical segment [9]. Other
imaging modalities have been proven useful such as single photon
emission computed tomography (SPECT) myocardial perfusion
imaging. This modality usually presents with three distinct findings;
1) an increased apical tracer uptake, 2) a spade-like configuration
of the LV chamber, and 3) the ‘‘Solar Polar” map pattern. This
presentation may be seen in 75% of patients [10].
AHCM can be diagnosed accurately with Cardiac computed
tomography (CCT), and the cardiac anatomy, function and coronary
artery are also assessed simultaneously. CMR is a valuable tool for
diagnosing this entity and it tends to be the modality of choice
when echocardiography produces suboptimal images. Furthermore,
CMR can accurately measure the myocardial wall thickness, the left
ventricular mass, the left atrial volume, and also search for apical
aneurysms and myocardial scarring/fibrosis [2].
Typical features of apical HCM include: audible and palpable
fourth heart sound, indicating LV relaxation impairment, large
negative T waves on EKG particularly in the left precordial leads,
“Spade-like” configuration of the left ventricular cavity at enddiastole
on imaging, and associated apical wall motion abnormalities
which may include hypokinesis and aneurysm formation [3].
Treatment
Regarding management, AHCM are treated like most HCM
patients. Symptomatic patients are primarily treated medically
that include the use of beta blockers, nondihydropyridine calcium
channel blockers as well as antiarrhythmics including Amiodarone
and Procainamide [4].
Beta-blockers have empirically been administered to AHCM
patients. Numerous studies investigated the effects of beta-blockers
on AHCM patients and reported symptom improvement. The
beneficial effects might be driven by sympathetic suppression of
heart rate, ventricular contractility, and stiffness. However, the longterm
benefits of beta-blockers in HCM patients are still unknown.
Although a cohort study reported that massive doses of propranolol
improved symptoms and long-term survival, this is not standard
procedure. Kim et al found beta-blocker use was significantly
associated with lower overall death but not lower cardiac death.
In other studies, beta-blockers reduced left ventricular outflow
tract obstruction and ventricular tachyarrhythmia. It is difficult to
determine the benefits of beta-blockers in HCM patients because the
agent, dosage, and duration were not consistent in all patients. Kim et
al strongly suggested that observation without medical attention for
apical HCM patients might in fact be harmful. Further randomized
trials should be conducted [11].
Surgical septal wall reduction is rarely done as AHCM almost
never cause Left Ventricular outflow obstruction. Our patient is
among the few treated surgically. Risk stratification for ventricular
tachyarrhythmia and sudden cardiac death is the same as for other
patients with HCM, but apical HCM is typically associated with a low
risk and almost never requires Implantable Cardioverter Defibrillator
placement for primary prevention [12].
Prognosis
Apical HCM has better mortality prognosis than other forms of
HCM but it is still associated with a relatively high rates of significant
cardiac events. Apical hypertrophic cardiomyopathy in North
American patients is very similar to that in Japanese patients, is not
associated with sudden cardiac death and has a benign prognosis in
terms of cardiovascular mortality. However, one third of APHCM
patients may develop unfavorable clinical events and potentially
life-threatening complications, such as myocardial infarction,
arrhythmias, and stroke [3]. In one study where 105 patients with
AHCM, with a mean age of 41 years, were followed for close to 14
years, the total cardiovascular mortality rate was 1.9% and the overall
estimated survival was 95% at 15 years. The study also showed that
30% of patients experienced serious cardiac complication, most
commonly atrial fibrillation (12%) or myocardial infarction (10%)
[12,13].
Conclusion
We presented a rare case of AHCM which was treated surgically. Symptomatic patients are primarily treated medically mostly by Beta blocker. Very few patients may need surgical management. This made our patient among the very few who underwent surgical intervention due to outflow obstruction and severe bradycardia. Thus counseling and management of patients with APHCM should be planned and followed accordingly.
Acknowledgment
We are indebted to Dominic Robine, DO, FACC, Cardiologist at Mountain View Hospital, Las Vegas – Nevada, USA for his continuous support and advice. And last, our special thanks to the patient for her patience and volunteering for this study.
Author’s Contributions
Michael GebreYohannes Tedla was the resident physician following this patient. Dawit Kibru Worku reviewed the literature. Michael GebreYohannes Tedla drafted the manuscript. Michael GebreYohannes Tedla and Dawit Kibru Worku reviewed the literature and the manuscript. Both authors read and approved the final manuscript.
References
- AlbanesiFilho FM, Castier MB, Lopes AS, Ginefra P. [Is the apical hypertrophic cardiomyopathy seen in one population in Rio de Janeiro city similar to that found in the East?]. Arq Bras Cardiol. 1997;69:117-123.
- Peter Rodgers-Fischl, Andrew RKolodziej, Vincent L Sorrell, Sarah SRugg. Apical Hypertrophic Cardiomyopathy in an African American: A Case Presentation and Literature Review. J CardiolClin Res. 2016; 4: 1056.
- Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am CollCardiol. 2002; 39:638-645.
- Maron MS, Finley JJ, Bos JM, Hauser TH, Manning WJ, Haas TS, et al. Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. 2008; 118:1541-1549.
- Raymond F. Stainback. Apical Hypertrophic Cardiomyopathy. Tex Heart Inst J. 2012; 39: 747-749.
- Syed Wamique Yusuf, Jaya D Bathina, Jose Banchs, Elie N Mouhayar, Iyad N Daher. Apical hypertrophic cardiomyopathy World J Cardiol. 2011; 3: 256-259.
- Kitaoka H, Doi Y, Casey SA, Hitomi N, Furuno T, Maron BJ. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003;92: 1183-1186.
- Karan Kapoor, AmalChaudhry, Matthew C Evans, Amish Sura. Apical Hypertrophic Cardiomyopathy Among Non-Asians: A Case Series and Review of the Literature. Cardiol Res. 2016; 7:46-50.
- Patel J, Michaels J, Mieres J, Kort S, Mangion JR. Echocardiographic diagnosis of apical hypertrophic cardiomyopathy with optison contrast. Echocardiography. 2002; 19: 521-524.
- Cianciulli TF, Saccheri MC, Masoli OH, Redruello MF, Lax JA, Morita LA, et al.Myocardial perfusion SPECT in the diagnosis of apical hypertrophic cardiomyopathy. J NuclCardiol. 2009; 16:391-395.
- Kim SH1, Kim SO, Han S, Hwang KW, Lee CW, Nam GB, et al. Long-term comparison of apical versus asymmetric hypertrophic cardiomyopathy. Int Heart J. 2013;54:207-211.
- Malik R, Maron MS, Rastegar H, Pandian NG. Hypertrophic cardiomyopathy with right ventricular outflow tract and left ventricular intracavitary obstruction. Echocardiography. 2014; 31:682-685.
- Mozaffarian D, Caldwell JH. Right ventricular involvement in hypertrophic cardiomyopathy: a case report and literature review. ClinCardiol. 2001; 24:2-8.