Case Report
Acute and Chronic Magnetic Resonance Imaging (MRI) Head Scan Changes in Adult Hyperammonemic Encephalopathy: A Case Report
Angela Wabulya1* and Scott Newsome2
1Department of Neurology, University of North Carolina, USA
2Department of Neurology, The Johns Hopkins University, USA
*Corresponding author: Angela Wabulya, Department of Neurology, University of North Carolina, 170 Manning Drive POB CB# 7025 Chapel Hill, NC, USA
Published: 20 Mar, 2017
Cite this article as: Wabulya A, Newsome S. Acute and
Chronic Magnetic Resonance Imaging
(MRI) Head Scan Changes in Adult
Hyperammonemic Encephalopathy: A
Case Report. Ann Clin Case Rep. 2017;
2: 1309.
ISSN: 2474-1655.
Abstract
Hyperammonemic encephalopathy is a type of an acute toxic-metabolic encephalopathy resulting from elevated plasma ammonia levels. Increased ammonia may be due to a variety of etiologies, at times leading to acute and chronic brain changes. Acute and chronic clinical and radiographic changes are well documented in the pediatric population, however, there is a paucity of reports pertaining to chronic changes in the adult population. In some adult patients, complete resolution of acute changes or mild cerebral atrophy has been reported differing from the pediatric population where significant chronic changes including neurologic devastation have been documented. Furthermore, hyperammonemia in adults is often attributed to hepatic disorders or typically thought of triggers such as use of Depakote, whose absence may at times be associated with late detection. We present a patient with acute hyperammonemic encephalopathy secondary to newly diagnosed Ornithine Transcarbamylase (OTC) deficiency followed over a3-year period with persistent cognitive dysfunction and significant Magnetic Resonance Imaging (MRI) of the head abnormalities. Hyperammonemic encephalopathy should be confirmed by serologic testing, however, in the event, that brain imaging is obtained prior to this, certain features of MRI of head should raise one’s suspicion and prompt early diagnosis and management. This case will emphasize the need for prompt diagnosis, identification of the causes and treatment of acute hyperammonemic encephalopathy especially in the absence of commonly implicated causes, to mitigate further brain injury as well as hopefully avoiding a recurrence.
Case Presentation
A 39-year-old man with no notable past medical history was admitted with altered mental status. His brother had died at 14 years of age following a confusion episode of an unclear etiology, while his sister was in good health. At the time of admission, he was married with two healthy sons and worked full-time as an operations manager with no limitations. Two weeks prior to admission, following a sick contact, he developed a sore throat, and cough. Initially not responding to cefuroxime, the fever and sore throat resolved with dexamethasone with clindamycin, however, four days prior to admission, he became progressively tired, spending long hours in bed, “fuzzyheaded”, confused with word finding difficulty and poor memory of recent events. At the local emergency room, he was found to be sleepier with paucity of movements prior to becoming unresponsive with no focal weakness. Initial work up including basic blood work, a head computed tomography (CT) scan, and lumbar puncture for cerebral spinal fluid analysis (CSF) were unrevealing. He was transferred to a tertiary center for further evaluation. At the tertiary institution, his vital signs were stable with a temperature of 36.1°C, blood pressure 130/40 mm Hg, heart rate 70 beats per minute, respiratory rate 12 breaths per minute and oxygen saturation of 100% on room air. He was not able to follow commands, produce spontaneous or provoked speech, although, had intermittent extremity movements, opening eyes unprompted and to noxious stimuli. There were no signs of meningeal irritation. Cranial nerves, motor and reflexes evaluation did not show any focal abnormalities. He was believed to have encephalopathy of an unclear etiology. His work up including, a complete blood count (CBC), comprehensive metabolic panel (CMP), toxicology, urinalysis showed elevated platelets 54110×9/L, ammonia 249 umol/L, relatively normal liver function tests alkaline phosphatase 59 U/L, aspartate transaminase (AST) 17 U/L, alanine transaminase (ALT) 41 U/L, AST/ALT ratio 0.4, albumin 4.3 g/dl, total bilirubin 0.7 mg/dl, and slightly elevated prothrombin time 14.4 seconds and international normalized ratio (INR) 1.4. A head CT scan showed effacement of the sulci, without intracranial hemorrhage or masses. The electroencephalogram showed diffuse delta rhythm without seizures. CSF analysis showed: White blood cell count (WBC): 2/2, Red blood cell counts (RBC): 78/43, Glucose 88 mg/dL (serum 220 mg/dl), Protein 19 mg/dL, no organisms seen on gram stain. Virology, cytopathology and bacterial culture were negative. The initial MRI of the head showed diffuse T2 weighted FLAIR (fluid attenuated inversion recovery) hyper intensities involving the frontal lobes, temporal lobes, and insular cortex (Figure 1A). At this point, the patient’s altered mental state was attributed to the elevated ammonia of an unknown etiology. He was maintained on a non-catabolic state by providing plenty of energy as dextrose and insulin as needed. Repeat ammonia level 11 hours after the initial test was 364 umol/L and 438 umol/L3 hours later with metabolic alkalosis: 7.45◊7.55. He had alow uric acid level at 2.7 mg/dL, normal acylcarnitine profile and plasma amino acids while the urine organic acids testing showed an orotic acid level peak. The allopurinol test demonstrated a 100-fold increase in the patient’s orotic acid level which together with the isolated increase in urine orotic acid, hyperammonemia, normal or low citrulline and metabolic alkalosis was suggestive of mild ornithine transcarbamylase (OTC) deficiency. The patient was managed in the neurology intensive unit with close monitoring of intracranial pressure and underwent dialysis in addition to administration of sodium benzoate/sodium and phenylacetate with 10% arginine HCL to help with stimulating alternate pathways of nitrogen excretion. Over a couple of weeks, he improved and was transferred to rehabilitation where his ammonia levels were checked weekly was normal. He was maintained on 60 grams of protein, 2200 calories with restricted nitrogen intake in addition to Sodium Phenylbutyrate 20 grams daily and Citrulline powder 8 grams daily. The patient improved gradually to the point of carrying out activities of daily living, however, not to the pre-illness state. There was no pre-illness intelligent quotient but the patient could resume his prior employment. The patient had serial MRI head scans that demonstrated acute changes with interval development of diffuse cortical atrophy (Figures 1B-E).
Figure 1
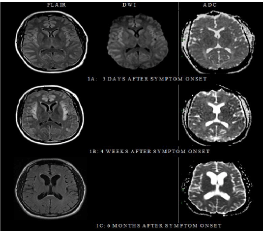
Figure 1
Brain MRI fluid attenuated inversion recovery (FLAIR), diffusion
weighted image (DWI), and apparent diffusion coefficient (ADC) sequences
respectively. (A) Three days after symptom onset; mild bilateral frontal and
insular cortex hyper intensities. (B) Four weeks later: Extensive cortical
signal abnormalities (restricted diffusion) maximal in the cingulated gyrus and
insular cortex. (C) Six months later; Resolution of the diffuse gyral swelling
with remnants of minimal FLAIR hyper intensity in the insular cortex with
accompanying volume loss as seen with enlargement of the Sylvain fissures
and the ventricular system. (D) One year and nine months later: Increased
ventricular size, reflecting increased cerebral volume loss. (E) Three years
and ten months later: Increased ventricular size in the frontal and temporal
horns of the lateral ventricles reflecting increased volume loss. FLAIR hyper
intensity in the insular regions and patchy supratentorial foci of hyper intense.
Discussion
Acute toxic-metabolic encephalopathy is an acute condition of global cerebral dysfunction in the absence of primary structural brain disease [1]. There are several causes of encephalopathy including elevated plasma ammonia levels. All forms of acute toxic-metabolic encephalopathy interfere with the function of the ascending reticular activating system and/or its projections to the cerebral cortex, leading to impairment of arousal and/or awareness [2]. Most toxic metabolic encephalopathies are reversible, making their prompt recognition and treatment important, however, certain metabolic encephalopathies, such as sustained hypoglycemia, thiamine deficiency (Wernicke's encephalopathy), may be associated with permanent structural brain damage [3]. Elevated ammonia levels can result from impaired endogenous hepatic detoxification (such as liver dysfunction, inborn urea cycle disorders and urea cycle suppressors like antiepileptic medications) or increased production of ammonia (such as overgrowth of urease-producing bacteria in the intestine or urinary tract). Most often in the adult population, liver dysfunction is implicated, however, as previously published other etiologies need to be considered especially in the absence of liver disease [4,5]. Prior publications show that ammonium ions penetrate the brain, reacting with astroglia-specific enzyme, glutamine synthetase to form glutamine whose osmotic action appears to be responsible for brain edema, intracranial hypertension, and cerebral hypoperfusion, particularly affecting the cingulate gyrus and insular cortex and laminar necrosis leading to variable symptomatology [6-8]. These changes may lead to stroke-like abnormalities in acute stages involving the cortex, especially the cingulate gyrus and insular cortex seen on MRI head scans, however permanent impairment, can occur [9,10]. It is therefore crucial to diagnose encephalopathy and its etiologies for optimal management and prevent acute and long term organ injuries [11]. The presentation of hyperammonemic states varies and is often nonspecific with clinical presentation including nausea, vomiting, protein intolerance, behavior changes, lethargy, ataxia, seizures, and coma typically presenting and precipitated by catabolic stress, infections, dehydration, protein load, surgery, childbirth, and gastrointestinal bleeding. To identify hyperammonemic states one must test for ammonia levels. In our patient, checking and following ammonia levels was vital for proper treatment. Whereas ammonia is easily checked in patients with typical etiologies such as liver disease or use of certain medications, it’s vital that health care providers consider checking ammonia in unexplained encephalopathic patients. In situations where brain imaging studies may have been obtained first, one has to be able to recognize patterns suggestive of hyperammonemic encephalopathy. In our patient, the initial head CT scan imaged showed sulcal effacement, raising concerns for increased intracranial pressure. MRI scans of the patient’s head later showed cortical signal abnormalities (restricted diffusion) maximal in the cingulated gyrus and insular cortex (Figure 1A) features previously reported [9,10] in hyperammonemia related encephalopathy. Recognition of such patterns should prompt health care providers to consider hyperammonemic encephalopathy and pursue prompt appropriate testing and management. Elevated ammonia levels in encephalopathic patients should prompt timely intervention to enhance its clearance w/wo decreasing production while identifying the causative factor to help with long term management and avoiding a recurrence. A systematic evaluation including consideration of genetic etiologies in the adult population has been previously published [4]. Particular attention should be paid to the history of previous episodes and to factors that might predispose to elevated ammonia or a family history of similar symptoms. In our patient, the encephalopathy of unclear etiology preceding his brother’s death, and the preceding infectious process in the patient were concerning. Further testing later showed features consistent with OTC, a not so uncommon disorder seen in adults [10]. Long-term complications in pediatric patient including learning disabilities with and without focal neurologic deficits as well as imaging evidence of acute or chronic ischemic cerebral damage which may be generalized, regional, or focal have been reported in hyperammonemic encephalopathies [10,12]. In the adult population, little is known about the long-term effects in adults with previous reports showing complete resolution of cortical lesions seen on MRI brain scans [6], while other cases noted mild atrophy in the cingulate gyrus and/or insular cortex [9]. Our patient responded to the appropriate management although never returned to his baseline. Follow-up patient MRI headscans (Figure 1B-E) showed diffuse cortical and subcortical gliotic and atrophic changes associated with CSF ventricular and extracerebral space enlargement. Extrapolation of Evan’s ratio of baseline to MRI head scans obtained at 21 months showed an increase from 0.21 to 0.42. It’s believed that the chronic changes are secondary to the extent of injury seen in the acute stage of hyperammonemic encephalopathy. These changes may likely correspond to the clinical deficits seen.
Conclusion
In conclusion, in patients of any age with altered consciousness, coma, or seizures with no clear anatomic or toxicologic cause, one should consider obtaining plasma ammonia levels. Elevated ammonia without evidence of hepatic failure or typically implicated etiologies should prompt more detailed work up including genetic etiologies. The authors encourage checking for ammonia levels in unexplained encephalopathy, in addition to recognizing brain imaging patterns previously reported in patients with hyperammonemic encephalopathy to enable prompt diagnosis and management to help mitigate potential subsequent morbidity and mortality.
References
- Chen R, Young GB. Metabolic Encephalopathies. In: Bolton CF, Young GB, editors. Baillere's Clinical Neurology. London: Balliere Tindall; 1996. 577.
- Plum F, Posner JB. The Diagnosis of Stupor and Coma. Philadelphia; FA Davis Company; 1982. p.177.
- Julio AC, Scott EK. Acute toxic metabolic encephalopathies in adults. Up To Date. 2016.
- Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest. 2007;132(4):1368-78.
- Hawkes ND, Thomas GA, Jurewicz A, Williams OM, Hillier CE, McQueen IN, et al. Non-hepatic hyperammonaemia: an important, potentially reversible cause of encephalopathy. Postgrad Med J. 2001;77(913):717-22.
- Chen YF, Huang YC, Liu HM, Hwu WL. MRI in a case of adult-onset citrullinemia. Neuroradiology. 2001;43(10):845-7.
- Choi JM, Kim YH, Roh SY. Acute hepatic encephalopathy presenting as cortical laminar necrosis: case report. Korean J Radiol. 2013;14(2):324-28.
- Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited disease. 8th ed. New York: McGraw-Hill: 1909-1963; 2001.
- U-King-Im JM, Yu E, Bartlett E, Soobrah R, Kucharczyk W. Acute Hyperammonemic Encephalopathy in Adults: Imaging Findings. Am J Neuroradiol. 2011;32(2):413-8.
- Bajaj SK, Kurlemann G, Schuierer G, Peters PE. CT and MRI in a girl with late-onset ornithine transcarbamylase deficiency: case report. Neuroradiol. 1996;38(8):796-9.
- Dasarathy S, Mookerjee RP, Rackayova V, Rangroo Thrane V, Vairappan B, et al. Ammonia toxicity: from head to toe?. Metab Brain Dis. 2017;32(2):529-38.
- CL Pridmore, JT Clarke, S Blaser. Ornithine Transcarbamylase Deficiency in Females: An Often Overlooked Cause of Treatable Encephalopathy. J Child Neurol. 1995;10(5):369-74.