Case Report
Thymic Carcinoid Tumor as Single Manifestation of Multiple Endocrine Neoplasia Type 1: A Clue to Suspect a Germline MEN1 Mutation?
Sergio Carrera1*, Intza Garin2, Aintzane Sancho1, Elena Beristain2, Eider Azkona1, Guillermo López-Vivanco1, Itziar Rubio1 and Cristina Martínez-Bouzas3
1Genetic Counseling Unit-Medical Oncology Department, University Hospital of Cruces, Spain
2Molecular Genetics Laboratory, University Hospital of Araba, Spain
3Molecular Genetics Laboratory, University Hospital of Cruces, Spain
*Corresponding author: Sergio Carrera, Genetic Counseling Unit-Medical Oncology Department, University Hospital of Cruces, Plaza de Cruces, S/N, 48903 Baracaldo, Vizcaya, Spain
Published: 09 Mar, 2017
Cite this article as: Carrera S, Garin I, Sancho A, Beristain
E, Azkona E, López-Vivanco G, Rubio I,
et al. Thymic Carcinoid Tumor as Single
Manifestation of Multiple Endocrine
Neoplasia Type 1: A Clue to Suspect
a Germline MEN1 Mutation?. Ann Clin
Case Rep. 2017; 2: 1293.
Abstract
Multiple endocrine neoplasia type 1 (MEN1) is a familiar tumor syndrome of endocrine neoplasia involving parathyroid, anterior pituitary and enteropancreatic neuroendocrine tissues. Thymic carcinoid tumors are rare tumors that occur in 2-8.2% of patients with MEN1 and they exhibit a predilection for men over women. Published data of thymic carcinoid tumors in MEN1 are scarce and they seldom constitute the first manifestation in this syndrome. We present a case of a woman with a thymic carcinoid tumor diagnosed at a very young age with no other clinical features of MEN1, and apparently not significant family history (mother with breast cancer developed at the age of 35), which was genetically diagnosed of a splice site mutation in MEN1 gene. Although no classical MEN1 clinical or familial criteria were met, the age of presentation of thymic carcinoid in our subject gave us the main clue of our clinical suspicion. Since MEN1 syndrome has a variable expression and it may not be exclusively correlated with endocrine tumors, we suggest that MEN1 mutational analysis should also be considered in all patients with carcinoid thymic tumors, especially if it is presented at young age, independently of the absence of classical clinical or familial MEN1 criteria.
Introduction
MEN1 which is also referred as Wermer syndrome is characterized by the combined occurrence
of tumors of the parathyroid glands, the pancreatic islet cells, and the anterior pituitary and
it is inherited in an autosomal dominant manner with high penetrance [1]. In addition to these
tumors, adrenal cortical tumors, carcinoid, facial angiofibromas, collagenomas and lipomatous
tumors have been described [2]. Parathyroid tumors, resulting in primary hyperparathyroidism
are the most common feature of MEN1 and occur in approximately 95% of patients [3]. Pancreatic
neuroendocrine tumors (NETs) occur in 40% and anterior pituitary tumors occur in 30% [4]. The
gene responsible for this syndrome is on chromosome 11q13 and encodes a 610 aminoacid protein,
menin, which has functions in cell division, genome stability and transcription regulation [5].
Different molecular genetic studies have confirmed the occurrence of de novo mutations of
the MEN1 gene in approximately 10% of patients with this syndrome [6]. A study has suggested
that near 70% of individuals with MEN1 currently die of causes directly related to MEN1 [7]; in particular, malignant pancreatic islet tumors and thymic carcinoid tumors are associated with a
marked increase in risk of death.
A diagnosis of MEN1 may be established in an individual by one of the following three
criteria: MEN1 may be clinically diagnosed in an individual on the basis of the occurrence of two
or more MEN1 associated endocrine tumors; familial MEN1 is defined as an individual who has
the occurrence of one of the MEN1 associated tumors and has a first degree relative with clinical
diagnosis of MEN1; also a genetic diagnosis of MEN1 is made on identification of a germline MEN1
mutation in an individual who may be asymptomatic [8].
Different germline MEN1 mutations have been described: approximately 23% are nonsense
mutations, 41% are frame shift deletions or insertions, 6% are in frame-deletions or insertions, 9% are splice-site mutations and 20% are missense mutations, and 1% are
whole o partial gene deletions [9].
The current guidelines recommend that MEN1 mutational
analysis should be performed in: 1) an index case with two or more
MEN1 typical associated endocrine tumors (parathyroid, pancreatic
or pituitary tumors), 2) asymptomatic first degree relative of a
known MEN1 mutation carrier, 3) a first degree relative of a MEN1
mutation carrier expressing familial MEN1 (having symptoms, signs,
biochemical or radiological evidence for one or more MEN1 associated
tumors) [10]. In addition MEN1 mutational analysis should be
performed in patients with suspicious or atypical MEN1: parathyroid
adenomas before the age of 30 years or multigland parathyroid
disease, gastrinoma and multiple pancreatic islet cells tumors any
age or individuals who have two or more MEN1 associated tumors,
which are not part of the classical triad of parathyroid, pancreatic islet
cell and anterior pituitary tumors [10].
Thymic carcinoids are rare neuroendocrine tumors. The
prevalence of thymic carcinoid tumors in patient with MEN1 ranged
from 2% to 8.2% in different series, and they exhibit a predilection
for men over women, with a male/female ratio of 20:1 [11]. With
scarce data reported in the literature of thymic carcinoids in patients
with MEN1, all available information of these tumors is relevant. A
retrospective study of MD Anderson Cancer Centre in 291 patients
who fulfilled clinical, genetic and or familial criteria for diagnosis of
MEN1 showed 9 cases (3.1%) of thymic carcinoids. The male/female
ratio was 2:1 and the mean age of diagnosis was 38.6 years. Almost
half of the patients already had distant metastases at the time of their
diagnosis [12].
Case Presentation
A 38 year old female was reported to our Medical Oncology
Genetic Counseling Department with diagnosis of thymic carcinoid
tumor. She was diagnosed at the age of 29. On December 2007
extended thymectomy was performed in spite of unresectable
macroscopic residual disease and the pathologist reported a
thymic classical carcinoid tumor. Cromogranine, sinaptofisine and
neuron specific enolase had strong positive staining and also weak
Follicle - Stimulating Hormone (FSH) staining was detected. Brain
magnetic resonance and body computed tomography scan did
not detect distant lesions. Blood analysis showed FSH increased
values in concordance with self-reported difficulty in conceiving.
Octreoscan revealed anterior mediastinal disease and treatment with
somatostatin analogues was initiated. Partial response was observed
until September 2013 when mediastinal mass increased in size and
left supraclavicular adenopathy and bone metastases were detected.
Everolimus treatment was added with good tolerability profile and
disease stabilization.
Our patient was the middle sister of three women. No relevant
clinical history was reported in her sisters. Her mother, 67 years old,
was diagnosed of breast cancer when she was 35 and she was under
clinical research of hypercalcemia when the patient was referred
to our Genetic Counseling Unit. The mother had only one sister,
who died at the age of 54 as a consequence of a brain hemorrhage.
Maternal grandmother died at advanced age (unspecified cause) and
maternal grandfather died in an accident when he was 40. No other
familial information was available.
Although no classical MEN1 clinical criteria were met, the age
of presentation of thymic carcinoid tumor in our subject with the
recently hypercalcemia detected in her mother gave us the main clue
of our clinical suspicion.
Figure 1
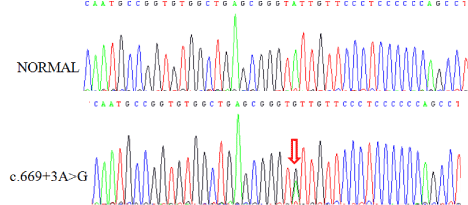
Figure 1
MEN1 germline analysis.
Methodology and Genetic Studies Results
Methodology
After written informed consent peripheral blood analysis were
collected from the patient. Mutation screening was performed on
genomic DNA, extracted by peripheral blood in EDTA, analyzing the
coding region (exon 2–10) and the exon-intron junctions (splicing
sites) of the MEN1 gene by PCR and Sanger sequencing (primers
and conditions available upon request). Obtained sequences were
compared to wild type reference sequence of the MEN1 gene, and
mutations were classified using the standard nomenclature for
the description of human DNA sequence variants. When a MEN1
mutation was detected, the mutation screening was extended to first
degree relatives of the proband, independently of the presence of
specific MEN1-related signs and symptoms.
Results
MEN1 germline analysis showed a substitution of an adenine
for guanine, in splicing donor site of intron 3, c.669+3A>G (Figure
1). This change was previously reported by Hai et al. [13] suggesting
that this variation affects splicing between exon 3 and 4. RT-PCR
with primers derived from exons 2 and 6 showed an aberrant 631
base pair (bp) band in addition to the 736 bp wild-type band. Direct
sequencing of the aberrant band revealed that a cryptic splice site in the middle of exon 3 was used and 105 bp 3´half of exon 3 was spliced
out. Once the MEN1 mutation was identified, study was offered to her
first degree relatives, confirming that her 67 years old mother, under
clinical study of hypercalcemia at the moment that germline mutation
was detected, was also carrier of c.669+3A->G MEN1 mutation. The
patient’s oldest sister was also asymptomatic carrier of the germline
MEN1 mutation.
Discussion
The present study reports the case of a patient with thymic
carcinoid tumor at the age of 29, with no other features of classical
MEN1 and no previous familial criteria of suspicion, in which we
identified a germline mutation in MEN1 of maternal inheritance.
Thymic carcinoid tumors are generally a late manifestation of MEN1
syndrome and few are the cases reported of very young patients and
only thymic carcinoids, without any other MEN1 related diseases
[14]. Approximately 90% of individuals diagnosed with MEN1
syndrome have an affected parent. However, the family history may
appear to be insignificant because of difficulty recognizing the disease
in family members, early death before the onset of symptoms or late
onset of the disorder in affected parent. Also the penetrance of this
syndrome approaches 100% with increasing age and with a variable
expression [15].
The mother was diagnosed of breast cancer when she was 35 years
old and germline MEN1 analysis confirmed she was also carrier of
the described pathologic mutation. To the best of our knowledge
there have been few studies regarding the association between breast
cancer and MEN1 [16] and further studies are needed, but we cannot
exclude that the breast cancer described in the mother of our patient
could be related to this syndrome.
Germline analysis of MEN1 gene revealed a mutation in splicing
donor site of intron 3, c.669+3A>G, which is a known mutation of the
MEN1 gene associated with the syndrome. Clinical manifestations of
the patient and affected family members in our report are different
from those of other studies with the same germline mutation [17],
which is in concordance with the absence of a MEN1 genotypephenotype
correlation [18].
The incidence of thymic carcinoids in patients with MEN1 has
been reported to be 3.6-8.4% and 25% of all thymic carcinoids occur
in patients with MEN1 [19]. The age of presentation of a thymic
carcinoid in different series is between 30 and 50 years [11]. Different
guidelines describe that MEN1 germline mutational analysis should
be considered in those presenting at an early age with a single,
apparently sporadic MEN1 associated tumor [20]. We suggest that
MEN1 mutational analysis should also be considered in all patients
with carcinoid thymic tumors, especially if early onset presentation,
regardless of the presence or absence of other clinical or familial
MEN1 features.
References
- Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. 1954; 16: 363-371.
- Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumours. Genet Med. 2009; 11: 825-835.
- Benson L, Ljunghall S, Akerstrom G, Oberg K. Hyperparathyroidism presenting as the first lesion in multiple endocrine neoplasia type 1. Am J Med. 1987; 82: 731-737.
- Calender A, Giraud S, Lenoir GM, Cougard P, Chanson P, Proye C. Hereditary multiple endocrine neoplasia. New genetic data and clinical applications in type 1 multiple endocrine neoplasia. Presse Med. 1995; 24: 542-546.
- Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997; 6: 1177- 1183.
- Bassett JH, Forbes SA, Pannett AA, Lloyd SE, Christie PT, Wooding C. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet. 1998; 62: 232-244.
- Goudet P, Murat A, Binquet C, Cardot-Bauters C, Costa A , Ruszniewki P, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d`Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg. 2010; 34: 249-255.
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1). J Clin Endocrinol Metab. 2012; 97: 2990-3011.
- Concolino P, Costella A, Capoluongo E. Multiple endocrine neoplasia type 1 (MEN1): an update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016; 209: 36-41.
- Newey PJ, Thakker R. Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract. 2011; 17: 8-17.
- Singh Ospina N, Maraka S, Montori V, Thompson GB, Young WF Jr. When and how should patients with multiple endocrine neoplasia type 1 be screened for thymic and bronchial carcinoid tumours? Clin Endocrinol (Oxf). 2016; 84: 13-16.
- Christakis I, Qiu W, Silva Figueroa AM, Hyde S, Cote GJ, Busaidy NL, et al. Clinical features, treatments, and outcomes of patients with thymic carcinoids and Multiple Endocrine Neoplasia Type 1 Syndrome at MD Anderson Cancer Center. Horm Canc. 2016; 7: 279-287.
- Hai N, Aoki N, Shimatsu A, Mori T, Kosugi S. Clinical features of multiple endocrine neoplasia type 1 (MEN1) phenocopy without germline MEN1 gene mutations: analysis of 20 Japanese sporadic cases with MEN1. Clin Endocrinol (Oxf). 2000; 52: 509-518.
- Gibril F, Chen YJ, Schrump DS, Vortmeyer A, Zhuang Z, Lubensky IA, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1 . J Clin Endocrinol Metab. 2003; 88: 1066-1081.
- Machens A, Schaaf L, Karges W, Frank-Raue K, Bartsch DK, Rothmund M, et al. Age related penetrance of endocrine tumors in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol. 2007; 67: 613-622.
- Jeong YJ, Kyu Oh H, Gu Bong J. Multiple endocrine neoplasia type 1 associated with breast cancer: a case report and review of the literature. Oncol Lett. 2014; 8: 230-234.
- Lim Park H, Ryung Yoo I, Hoon Kim S, Lee S. Multiple endocrine neoplasia type 1 with anterior mediastinal parathyroid adenoma: successful localization using Tc-99m sestamibi SPECT/CT. Ann Surg Treat Res. 2016; 91: 323-326.
- Lim LC, Tan MH, Eng C, Teh BT, Rajasoorya RC. Thymic carcinoid in multiple endocrine neoplasia 1: genotype-phenotype correlation and prevention. J Intern Med. 2006; 259: 428-432.
- Sakurai A, et al. Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clin Endocrinol (Oxf). 2012; 76: 533-539.
- Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 2014; 386: 2-15.