Case Report
Aggressive Angiomyxoma of the Abdominopelvic Cavity: A Case Report
Yun-jie Tian, Shan Kang*, Li Li and Xi-wa Zhao
Department of Obstetrics and Gynecology, The Fourth Hospital of Hebei Medical University, PR China
*Corresponding author: Shan Kang, Department of Obstetrics and Gynecology, The Fourth Hospital of Hebei Medical University, 12 Jiankang Road, Shijiazhuang 050011, PR China
Published: 08 Feb, 2017
Cite this article as: Yun-jie Tian, Kang S, Li L, Xi-wa
Zhao. Aggressive Angiomyxoma of the
Abdominopelvic Cavity: A Case Report.
Ann Clin Case Rep. 2017; 2: 1263.
Abstract
Background: Aggressive Angiomyxoma (AAM) is a rare type of mesenchymal tumor that affects the
pelvic and perineal regions of premenopausal women. AAM is a slow-growing, locally infiltrating
tumor that tends to recur locally but is unlikely to metastasize. To date, surgery is the standard
treatment for AAM, although it carries limited success rates. The literature around AAM is also
limited.
Case Presentation: We report an unusual case of a 67-year-old woman with an extensive soft-tissue
mass in the pelvic and abdominal cavities. We performed complete tumor excision, and a diagnosis
of AAM was confirmed via pathology. The mass recurred within 5 months of surgery and grew in
size rapidly. A wider tumor excision was performed 8 months after the initial surgery; however,
a recurrent mass was again detected during the subsequent follow-up period. Four months after
the second resection, the patient underwent 1 cycle of chemotherapy (cisplatin, ifosfamide, and
epirubicin) at another hospital; however, the diameter of the mass continued to increase. At our
last follow-up visit (15 months after the initial surgery), the patient was in poor health and had
abandoned further treatment.
Conclusion: AAM should be considered in the differential diagnosis of asymptomatic, slowgrowing
masses in the abdominopelvic cavity. Wide local excision and long-term follow-up are
essential for treating AAM.
Keywords: Aggressive angiomyxoma; Mesenchymal tumor; Wide local excision
Introduction
Aggressive angiomyxoma (AAM) was first described by Steeper and Rosai in 1983 as a distinct
mesenchymal tumor of the pelvis and perineum in women [1]; it was described as “aggressive” to
emphasize the neoplastic nature of the blood vessels and its locally infiltrative and recurrent nature.
Fewer than 250 cases of AAM have been reported in the literature, most of which occurred in
women [2]. A recent literature review that assessed over 100 cases of AAM calculated a female-tomale
incidence ratio of 6.6:1 [2]. Women who develop AAM are predominantly of childbearing age;
the peak incidence is in the fourth decade of life, although the age distribution (ranging from 6 to
77 years) is wide [1-6]. AAM occurs predominantly in the vulvovaginal, perineal, and groin regions;
those that occur in men involve analogous sites, including the inguinoscrotal region and perineum
[7].
Surgical excision is generally recommended for the management of AAM. In 1992, Simo et
al. [8] proposed that curative treatment for AAM should involve wide surgical excision based on precise histopathological diagnoses. Here, we report a rare case of a 67-year-old woman with AAM in the pelvic and abdominal cavities.
Case Presentation
A married 67-year-old woman (gravida 2; para 1) complained of progressive abdominal
distension and anorexia. Upon physical examination, her abdomen was soft and distended.
Gynecological examination revealed a 200 × 100 × 100 mm mass with low mobility and without
clear margins. Ultrasonography revealed an irregularly shaped soft-tissue tumor with mixed
echogenicity, approximately 195 × 129 × 108 mm in size (Figure 1). Computed Tomography (CT)
of the abdomen and pelvis revealed a huge, hypodense mass in the pelvic and lower abdominal
regions with a longitudinal diameter of 190 mm (Figure 2). The patient had a normal menstrual history, no history of taking drugs, and had not undergone any prior
surgical procedures. Surgical exploration revealed a soft, poorly
circumscribed mass, approximately 200×150×100 mm in size, within
the pelvis and abdomen. A spontaneous rupturing of the mass, which
produced bloody ascites approximately 200 mL in volume, was
also noted. The tumor had adhered to the intestines and the major
omentum, and covered the uterus as well as the adnexa bilaterally.
After careful dissection, the uterus and the adnexa appeared to be
normal. The surfaces of the liver, spleen, stomach, and appendix were
smooth. Owing to the high possibility of a mesenchymal tumor, we
performed tumor resection, total abdominal hysterectomy, bilateral
salpingo-oophorectomy, and omentectomy. On gross examination,
there were large, poorly circumscribed lesions with irregular
extensions into the surrounding tissues. Microscopic examination
showed the proliferation of small fusiform or star-shaped cells
without cytonuclear atypia that were interspersed in a myxoid
background enclosing several capillary structures with thin walls
(Figure 3). Immunohistochemistry showed that the tumor strongly
expressed desmin and moderately expressed vimentin, CD99,
CD34, and smooth muscle actin; the tumor was negative for CD117,
cytokeratin, estrogen receptor, progesterone receptor, and S100. The
diagnosis of AAM was confirmed on pathologic examination. After
radical surgery, no further treatment was conducted except for close
follow-up.
Five months after surgery, a recurrence of the mass was detected.
Ultrasonography revealed a hypoechoic, solid tumor (71 × 56 × 42
mm) located on the vaginal stump. At the next monthly follow-up,
magnetic resonance imaging (MRI) confirmed an irregularly shaped
tumor in the pelvic cavity, measuring 84 mm in diameter (Figure 4).
Eight months after the surgery, the recurrent mass was approximately 204 × 113 × 97 mm in size; the mass grew gradually to the point where
it severely affected gastrointestinal peristalsis and caused abdominal
distention. Hence, the patient underwent a second, wide excision.
Surgical exploration found that the tumor in the pelvis and abdomen,
200 × 185 × 160 mm in size, was gelatinous, poorly circumscribed,
and had no capsule. The recurrent tumor had occupied the interintestinal
space and the vaginal stump. After the second operation,
we continued close follow-up. Four months after the second tumor
resection, a third recurrent mass developed. Ultrasonography
revealed a solid, cyst-like mass in the pelvis and abdomen 294 ×
272 × 100 mm in size. Following the third recurrence, the patient
sought treatment at another hospital, where she was administered
a chemotherapy regimen of cisplatin, ifosfamide, and epirubicin for
1 cycle. Five months after the second operation, ultrasonography
indicated an irregularly shaped mass measuring 409 × 335 × 128 mm.
When she was last seen 15 months after the initial surgery, the patient
could barely eat or sleep, and had abandoned treatment.
Figure 1
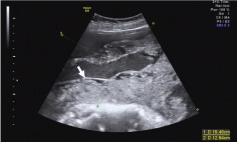
Figure 1
Transabdominal ultrasonography revealed an irregularly shaped,
soft-tissue tumor with mixed echogenicity (arrow).
Figure 2
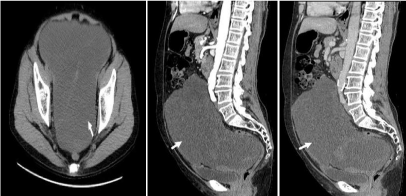
Figure 2
Computed tomography (CT) of the abdomen and pelvis revealed
a large, hypodense mass (arrows) in the pelvic and lower abdominal regions
before the first surgery. (A) plain CT scan; (B) CT scan in the arterial phase;
(C) CT scan in the venous phase.
Figure 3
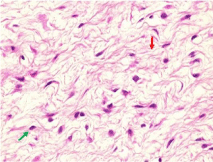
Figure 3
Photomicrograph (hematoxylin-eosin stain, original magnification
×40) showing the proliferation of small fusiform or star-shaped cells without
cytonuclear atypia (green arrow) interspersed in a myxoid background (red
arrow) enclosing several capillary structures with thin walls.
Figure 4
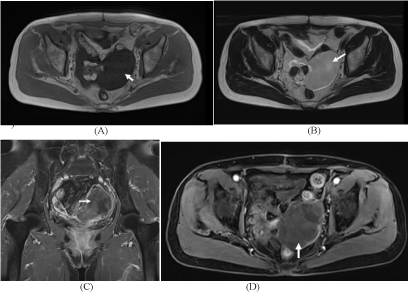
Figure 4
Magnetic resonance imaging (MRI) at first recurrence confirmed an
irregularly shaped tumor in the pelvic cavity measuring 84 mm in diameter.
(A) Axial unenhanced T1-weighted MRI showed an aggressive angiomyxoma
(arrow) that was isointense to the muscle; (B) Axial unenhanced T2-weighted
MRI showed that the tumor was hyperintense relative to the muscle, and a
swirling or layered appearance (arrow) was noted within the mass; (C) The
tumor (arrow) was isointense on coronal enhanced T1-weighted imaging;
(D) An axial enhanced T1-weighted image showed the tumor (arrow) with
heterogeneous enhancement.
Discussion
AAM is one of the rarest and perhaps most misdiagnosed types
of genital stromal tumors. It is often detected incidentally during
physical examinations, owing to its slow growth pattern and lack of
symptoms. If they manifest, symptoms may include pelvic fullness
and pressure, dysmenorrhea, and changes in bowel and bladder
function [3,9-11]. No characteristic symptoms of AAM have been
identified, making it difficult to diagnose this disease.
The appearance of AAM on CT is variable. It may be a welldefined,
homogeneous mass that is hypodense relative to muscle,
or may be predominantly cystic with solid components [12].
Characteristic appearances on MRI include hypointensity on T1-
weighted images and hyperintensity on T2-weighted images. The
“swirl sign” is characteristic on T2-weighted MRI [13]. With its better
soft tissue resolution, MRI is more reliable for the diagnosis of AAM.
MRI was performed following the first recurrence of the tumor in
our patient to evaluate her condition; however, CT was used during
follow-up visits owing to the patient’s financial situation. Given its
characteristic features, MRI should be used for delineating the extent
of disease, determining eligibility for surgery and clinical follow-up
examinations for recurrent tumors if a patient’s financial situation
allows. On gross examination, AAMs are typically soft, bulky masses.
Microscopic examination shows a sparsely cellular tumor composed
of pale-to-eosinophilic stroma with numerous haphazardly arranged
blood vessels that stand out against a myxoid background. There is
no specific immunohistochemistry marker for AAM; however, these
tumors are generally positive for vimentin and desmin.
The preferred treatment for AAM is surgery, although achieving
negative resection margins is challenging owing to the infiltrative
nature of the tumor [14]. It is especially difficult to remove large and
deep-seated tumors in the pelvis compared to the small and superficial
tumors of the vulva or vagina. To assess the necessity of radical
resections, researchers analyzed data from 111 patients with AAM
and found no statistical differences in survival according to the time
to relapse or resection margin status [2]. Although complete surgical
resection should be the preferred aim, incomplete or partial resection
is acceptable when significant operative morbidity is anticipated or
when the preservation of fertility is required. Our patient experienced
recurrence despite wide excision with negative surgical margins; she
chose not to undergo a third excision after recurrence and sought
chemotherapy at another hospital. Generally, adjuvant radiotherapy
or chemotherapy is not a preferred route because of the low mitotic
activity of AAM [3]; however, several cases of tumor recurrence have
been treated with radiation therapy resulting in patients achieving
relapse-free intervals of 2–3 years [15,16]. To our knowledge, ours
is the first report of chemotherapy used for a patient with AAM;
however, it did not provide any curative benefit.
The treatment of recurrences, however, may be challenging and
require various therapeutic options to be explored. This is especially
true given that no single modality has been proven to be effective [17].
AAM tends to recur after surgical excision despite being a benign and
non-invasive tumor; such recurrence can occur at the original site
after the initial resection [18]. Recurrence rates range from 25% to
47%, and 85% of all recurrences appear within 5 years of initial surgery
[2,3,19]. It is highly recommended that patients with AAM be closely
followed by using clinical and imaging tests, particularly MRI. To our
knowledge, only 2 cases of metastasis from AAM have been reported
in the literature [20,21], suggesting that metastasis is an exceedingly rare event. Because recurrences have been observed 14 years after the
initial diagnosis [14], proper management, combined with long-term
follow-up, is necessary to identify recurrences of AAM.
Conclusion
AAM should be considered in the differential diagnosis of a painless, soft, slow-growing mass in women. Wide local excision remains the first-line treatment to date. Long-term follow-up and careful monitoring with imaging techniques are required for timely identification of the recurrent tumors and prompt resection. In our patient, chemotherapy did not provide any curative benefit.
References
- Steeper TA, Rosai J. Aggressive angiomyxoma of the female pelvis and perineum. Report of nine cases of a distinctive type of gynecologic softtissue neoplasm. Am J Surg Pathol. 1983; 7: 463-475.
- Chan YM, Hon E, Ngai SW, Ng TY, Wong LC. Aggressive angiomyxoma in females: is radical resection the only option?. Acta Obstet Gynecol Scand. 2000; 79: 216–220.
- Fetsch JF, Laskin WB, Lefkowitz M, Kindblom LG, Meis-Kindblom JM. Aggressive angiomyxoma: a clinicopathologic study of 29 female patients. Cancer. 1996; 78: 79-90.
- Piura B, Shaco-Levy R. Pedunculated aggressive angiomyxoma arising from the vaginal suburethral area: case report and review of literature. Eur J Gynaecol Oncol. 2005; 26: 568-571.
- Chihara Y, Fujimoto K, Takada S, Hirayama A, Cho M, Yoshida K, et al. Aggressive angiomyxoma in the scrotum expressing androgen and progesterone receptors. Int J Urol. 2003; 10: 672-675.
- Baruah S, Latthe P, Bhatti NR, Ghataura SS. Aggressive angiomyxoma of the vulva. Hosp Med. 2004; 65: 248–249.
- Iezzoni JC, Fechner RE, Wong LS, Rosai J. Aggressive angiomyxoma in males. A report of four cases. Am J Clin Pathol. 1995; 104: 391-396.
- Simó M, Zapata C, Esquius J, Domingo J. Aggressive angiomyxoma of the female pelvis and perineum. Report of two cases and review of the literature. Br J Obstet Gynaecol. 1992; 99: 925-927.
- Nakamura T, Miura K, Maruo Y, Sunayama K, Maruyama K, Kashiwabara H, et al. Aggressive angiomyxoma of the perineum originating from the rectal wall. J Gastroenterol. 2002; 37: 303-308.
- Magtibay PM, Salmon Z, Keeney GL, Podratz KC. Aggressive angiomyxoma of the female pelvis and perineum: a case series. Int J Gynecol Cancer. 2006; 16: 396-401.
- Nyam DC, Pemberton JH. Large aggressive angiomyxoma of the perineum and pelvis: an alternative approach. Report of a case. Dis Colon Rectum. 1998; 41: 514–516.
- Jeyadevan NN, Sohaib SA, Thomas JM, Jeyarajah A, Shepherd JH, Fisher C. Imaging features of aggressive angiomyxoma. Clin Radiol. 2003; 58: 157-162.
- Srinivasan S, Krishnan V, Ali SZ, Chidambaranathan N. "Swirl sign" of aggressive angiomyxoma-a lesser known diagnostic sign. Clin Imaging. 2014; 38: 751-754.
- Haldar K, Martinek IE, Kehoe S. Aggressive angiomyxoma: a case series and literature review. Eur J Surg Oncol. 2010; 36: 335-339.
- Rhomberg W, Jasarevic Z, Alton R, Kompatscher P, Beer G, Breitfellner G. Aggressive angiomyxoma: irradiation for recurrent disease. Strahlenther Onkol. 2000; 176: 324-326.
- Suleiman M, Duc C, Ritz S, Bieri S. Pelvic excision of large aggressive angiomyxoma in a woman: irradiation for recurrent disease. Int J Gynecol Cancer. 2006; 16: 356-360.
- Kura MM, Jindal SR, Khemani UN. Aggressive angiomyxoma of the vulva: An uncommon entity. Indian Dermatol Online J. 2012; 3: 128-130.
- Dierickx I, Deraedt K, Poppe W, Verguts J. Aggressive angiomyxoma of the vulva: a case report and review of literature. Arch Gynecol Obstet. 2008; 277: 483-487.
- Granter SR, Nucci MR, Fletcher CD. Aggressive angiomyxoma: reappraisal of its relationship to angiomyofibroblastoma in a series of 16 cases. Histopathology. 1997; 30: 3-10.
- Siassi RM, Papadopoulos T, Matzel KE. Metastasizing aggressive angiomyxoma. N Engl J Med. 1999; 341: 1772.
- Blandamura S, Cruz J, Faure Vergara L, Machado Puerto I, Ninfo V. Aggressive angiomyxoma: a second case of metastasis with patient's death. Hum Pathol. 2003; 34: 1072-1074.