Case Report
Diabetic Foot Syndrome and Anaplerotic Therapy in a Long-Surviving Patient with Leprechaunism (Donohue Syndrome)
Bergis Dominik1*, Böhles Hans2 and Badenhoop Klaus1
1Division of Endocrinology and Diabetes, Department of Internal Medicine, Goethe-University Hospital, Germany
2Department of Pediatrics, Goethe-University Hospital, Germany
*Corresponding author: Dominik Bergis, Department of Internal Medicine 1, Division of Endocrinology & Diabetes, Goethe-University Hospital, Theodor-Stern-Kai 7, 60590 Frankfurt, Germany
Published: 02 Dec, 2016
Cite this article as: JDominik B, Hans B, Klaus B. Diabetic
Foot Syndrome and Anaplerotic
Therapy in a Long-Surviving Patient
with Leprechaunism (Donohue
Syndrome). Ann Clin Case Rep. 2016;
1: 1200.
Abstract
Background: Donohue syndrome (DS), also known as Leprechaunism, describes a rare form of
congenital insulin resistance associated with refractory hyperglycemia, hyperinsulinism and severe,
characteristic musculoskeletal deformities and facial dysmorphia. Prognosis of DS is poor and most
individuals die early in infancy.
Case Report: We report a long-surviving male patient with Donohue syndrome suffering from
diabetic foot lesions at the age of 13. Long survival was most likely in part related to anaplerotic
therapy containing aspartate and citrate – two citric acid cycle intermediate metabolites missing
in DS. When first seen in our outpatient clinic, the boy had several ulcers on the heel, the forefoot
and lower leg. Conventional treatment with off-loading, wound debridement and antibiotic therapy
was initiated. After 12 weeks, ulcers had resolved almost completely – despite insufficient glycemic
control.
Discussion: The nutritional concept of anaplerotic therapy has never been reported in DS before
and may be crucial in order to aim at prolonged survival as long as better treatment options are not
available. Long term diabetic complications such as diabetic foot syndrome have not been observed
in DS yet, but must be expected when patients survive infancy.
Keywords: Diabetic foot; Nutrition and diet; Neuropathy; Insulin sensitivity
Introduction
Donohue-syndrome (DS), first described by the Canadian pathologist William L. Donohue in
1948 [1], is the most severe form of an insulin-receptor defect with minimal or no residual function
[2]. DS is very rare with less than 1/1.000.000 births and due to homozygous or compoundheterozygous
mutations of the insulin-receptor gene located on the short arm of chromosome 19
(INSR 19p13.2). Several mutations of the gene have been discovered so far, including a nonsense
mutation causing a frame shift [3], a single missense mutation or a single codon change that altered
isoleucine to methionine in the receptor protein [4]. The mutation either causes malfunction of the
receptor´s auto-phosphorylation leading to a complete shutdown of the downstream signal cascade
or a conformational change in the alpha/beta-subunit leading to the inability of insulin binding to
its receptor [5].
Donohue syndrome, formerly referred to as leprechaunism, is initially characterized by severe
intrauterine growth restriction. The term Leprechaunism has largely been abandoned since it is
perceived by parents as stigmatizing. It was derived from the “Elfin” features that patients with
DS typically display: additional to post-natal growth retardation are protuberant and low-set ears,
flaring nostrils, and thick lips. Physical features comprise an enlarged clitoris in affected females,
and an enlarged penis in affected males. Additionally, lipoatrophy and muscular hypotrophy are
found as well as typical symptoms of insulin resistance, such as hirsutism and acanthosis nigricans.
Excessive hyperglycemia followed by phases of hypoglycemia together with severe hyperinsulinemia
is present in all patients with DS. Hyperglycemia does not respond to conventional antihyperglycemic
treatment, whereas insulin therapy or insulin sensitizers such as rosiglitazone are
nevertheless applied in those patients. Additionally, therapy with recombinant human insulin-like
growth factor I (IGF-I) is frequently established and provides – due to its homology with insulin and
its receptors - some glycemic control and acceleration of growth. However, prognosis of DS remains poor and most individuals die early in infancy.
Here we describe the first case of a 13-year old male patient with
DS who suffered from diabetic foot lesions, which were subjected
to conventional wound- and systemic treatment. Diabetic foot
syndrome as the most severe complication of diabetes mellitus and
has never been described in patients with DS before.
Case Presentation
The boy was delivered in April 2000 after 38 weeks of gestation
by caesarian section as the second twin of consanguineous Turkish
parents. Both the boy and his brother showed the clinical phenotype
of leprechaunism and DS was diagnosed according to clinical and
biochemical features. A molecular characterization of the underlying
mutation was not consented by the parents. Prenatal development of
the boy was characterized by severe intrauterine growth retardation,
which lead to a prolonged postnatal hospitalization of 8 weeks. From
early on he suffered from severe hyperglycemias refractory to insulin
treatment followed by phases of hypoglycemia. Blood glucose levels
were mostly between 2.8 and 27.8 mmol/l. Continuous nutrition
every 2 hours was necessary in order to avoid severe hypoglycemia.
At the age of 3 years, treatment with the PPAR gamma agonist
rosiglitazone was initiated at a dose of 4mg/d. However, glycemic
control was not achieved and the boy subsequently suffered from
recurrent urinary and respiratory infections, compelling a continuous
antibiotic treatment with trimethoprim and later erythromycin.
Another important feature in his first years was massive dental
caries, apart from other problems such as persistent severe growth
retardation and clinical signs of insulin resistance (hirsutism,
acanthosis nigricans). Meanwhile, the patients´ brother had died
at the age of 18 months due to cerebral hemorrhage after volatile
anesthesia. At the age of 4, the boy suffered from sepsis caused by
Pseudomonas aeruginosa after surgical treatment of an inguinal
hernia. During sepsis a severe ketoacidosis occurred. The absence of
citric acid cycle intermediate metabolites aspartate and succinate was
held responsible and subsequently, an anaplerotic therapy containing
aspartate and citrate together with a ketogenic diet was initiated.
Additionally, the boy received intravenous lipid nutrition and – later
on – 3-OH-butyrat orally. Upon these measures the patient’s clinical
condition significantly improved despite persisting hyperglycemia.
subsequently, all antidiabetic treatment was stopped. At the age of 13,
the boy developed clinical signs of a diabetic foot syndrome and was
referred to our Diabetes outpatient clinic. On admission, the growthrestricted
boy (height 97 cm, weight 13 kg) showed the typical clinical
appearance of DS. His physical condition was stable, blood pressure
was within normal range and he was afebrile. Peripheral pulses
were palpable, whereas neuropathy assessment was not possible
due to the boy´s mental retardation. He presented with three ulcers:
one lesion of about 3x1.5cm on the forefoot, stage IB according to
Wagner-Armstrong, one ulcer on the lower leg of about 4x4.5cm,
also stage IB and a deeper lesion of 3x3cm, Wagner-Armstrong stage IIB, on his right heel (Figure 1 a-c). Bacterial smears were
taken and showed a mixed flora of Klebsiella species, Enterococcus
faecalis and Staphylococcus aureus. We initiated antibiotic treatment
with amoxicillin/clavulanic acid and started standard wound care
consisting of cleansing and debriding the ulcers. The boy received a
heel-wedge off-loading shoe-cast and parents were advised to apply
it during daytime. Additionally, a dermatologist excluded livedo
vasculopathy on the lower leg by skin biopsy and histology.
After 4 weeks of treatment, the wound situation had substantially
improved (Figure 2 a-c) – despite the persistent hyperglycemia. After
a treatment period of 12 weeks, ulcers of the lower leg and the heel
were fully epithelialized and the ulcer of the forefoot was in complete
clinical remission (Figure 3 a-c). After that we lost the patient to
follow-up. He was only seen by his local pediatrician. One year later,
the boy succumbed to pneumonia.
Figure 1a-c
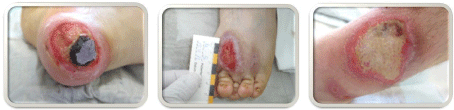
Figure 1a-c
Initial presentation of foot ulcers.
Figure 2a-c
Figure 2a-c
Clinical course of foot ulcers after 4 weeks.
Figure 3a-c
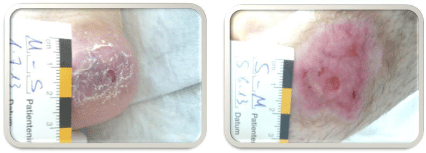
Figure 3a-c
Clinical course of foot ulcers after 12 weeks.
Discussion
Here we report the first case of a patient with Donohue syndrome
suffering from diabetic foot syndrome at the age of 13. Most
interestingly he showed a good response to conventional treatment
by immobilization, off-loading and anti-infective treatment despite
poor glycemic control due to the untreatable diabetes.
The boy´s unexpected long survival until the age of 13 is in
contrast to most DS patients who die within the first years of infancy.
We understand this is as likely success of an experimental nutritional
concept, that was established when the patient was 4 years old. Prior
to initiation of the nutritional concept, treatment with recombinant
human insulin-like growth factor I (IGF-I) as used in other DS
patients [6-8], had been considered but was not applied in the boy.
The nutritional concept was mainly based on the consideration of
a lacking cellular glucose uptake caused by the underlying insulin
receptor defect leading to glucagon-dependent lipolysis and free
fatty acid metabolization – a situation comparable to ketoacidosis
in type 1 diabetes. Since glucose is not available for the citric acid
cycle, its replenishment with necessary intermediates, the so called
anaplerosis, is impaired which leads to a diminished 3-hydroxybutyric
acid (3-OH-butyrate)/Acetoacetate-ratio, a reduced Aspartate
production and ultimately a rise of the lactate/pyruvate ratio. After
careful consideration of these biochemical pathways, an individual
therapeutic trial using 3-hydroxybutyric acid (3-OH-butyrate) as
an energy source was undertaken. Already after application of 5g
3-OH-butyrate a day, ketone bodies were undetectable in urine thus
implying that al 3-OH-butyrate was metabolized by the patient.
Additionally, substitution of the citric acid cycle in terms of an
anaplerotic therapy with its metabolites aspartate and succinate was
initiated in combination with a ketogenic diet. Antidiabetic treatment
was continued using 4mg of rosiglitazone and 2g of metformin.
During the clinical course, weight gain from less than 8.5kg
to 12kg was observed until the age of 6. Between 6 and 12 years,
consultations became less frequent, but when seen in our diabetic
foot outpatient clinic, the body weight had remained stable at 13kg.
We therefore conclude that an experimental nutritional concept
is crucial in patients with DS in order to aim at prolonged survival as
long as better treatment options are not available. Both anaplerotic
therapy and ketogenic diet have proven to be safe in children with
epileptic seizures [9,10] and can therefore be tried as therapeutic
options in DS.
Clinical decisions are challenging in DS not only because of the
untreatable molecular defect but also for ethical reasons when parents
are faced with the medical consequences of consanguinity. This report
shall widen the narrow window of therapeutic opportunities in DS.
References
- Donohue WL, Edwards H. Dysendocrinism. The Journal of Pediatrics. 32: 9.
- Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev. 2011; 32: 498-514.
- Psiachou H, Mitton S, Alaghband-Zadeh J, Hone J, Taylor SI, Sinclair L. Leprechaunism and homozygous nonsense mutation in the insulin receptor gene. Lancet; 1993: 342: 924.
- Longo N, Langley SD, Griffin LD, Elsas LJ. Two mutations in the insulin receptor gene of a patient with leprechaunism: application to prenatal diagnosis. J Clin Endocrinol Metab. 1995: 80: 1496-501.
- Falik Zaccai TC, Limor Kalfon, Aharon Klar, Mordechai Ben Elisha, Haggit Hurvitz, Galina Weingarten, et al. Two novel mutations identified in familial cases with Donohue syndrome. Mol Genet Genomic Med. 2014; 2: 64-72.
- Backeljauw PF, Alves C, Eidson M, Cleveland W, Underwood LE, et al. Effect of intravenous insulin-like growth factor I in two patients with leprechaunism. Pediatr Res. 1994; 36: 749-54.
- de Kerdanet M, Caron-Debarle M, Nivot S, Gaillot T, Lascols O, Fremont B, et al. Ten-year improvement of insulin resistance and growth with recombinant human insulin-like growth factor 1 in a patient with insulin receptor mutations resulting in leprechaunism. Diabetes Metab. 2015; 41: 331-337.
- Desbois-Mouthon C, Danan C, Amselem S, Blivet-Van Eggelpoel MJ, Sert-Langeron C, Goossens M, et al. Severe resistance to insulin and insulin-like growth factor-I in cells from a patient with leprechaunism as a result of two mutations in the tyrosine kinase domain of the insulin receptor. Metabolism. 1996; 45:1493-500.
- Kovac S AY, Abramov, MC Walker. Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology. 2013; 69: 96-104.
- Maiorana A, Lucilla Manganozzi, Fabrizio Barbetti, Silvia Bernabei, Giorgia Gallo, et al. Ketogenic diet in a patient with congenital hyperinsulinism: a novel approach to prevent brain damage. Orphanet J Rare Dis. 2015; 10: 120.