Case Report
Retinoblastoma and Second Bone Sarcomas: A Pediatric Case Report
Zied Jlalia*, Aymen Zaier, Samih Kacem and Mahmoud Smida
Kassab Orthopedics Institute, Medicine Faculty of Tunis, University of El Manar, Tunisia
*Corresponding author: Zied Jlalia, Kassab Orthopedics Institute, Medicine Faculty of Tunis, University of El Manar, Ksar Said 2010, La Manouba, Tunisia
Published: 27 Nov, 2016
Cite this article as: Jlalia Z, Zaier A, Kacem S, Smida M.
Retinoblastoma and Second Bone
Sarcomas: A Pediatric Case Report.
Ann Clin Case Rep. 2016; 1: 1190.
Abstract
The occurrence of secondary malignant neoplasia remains the most important long-term risk but rare of retinoblastoma. Osteosarcoma represents the most frequent second malignant tumor. We report here a case of a right femur osteosarcoma in a 7 years and half old-boy after being treated for retinoblastoma at two years of age. The treatment consisted of chemotherapy and conservative surgery with intercalary tumor resection and reconstruction by two autologous fibulas. Three years after, the child died from pleural metastasis. The purpose of this report is to review the genetic relationship between retinoblastoma and osteosarcoma and to discuss treatment protocol for the latter.
Keywords: Retinoblastoma; Second sarcoma; Osteosarcoma; Rbgene
Introduction
Retinoblastoma is the most common ocular primitive tumors [1]. The appearance of Secondary Malignant Neoplasia (SMN) remains the most important long-term risk but rare of retinoblastoma [1-3]. Generally, sarcomas are the most frequent of SMN, especially osteosarcoma, with uncertain prognosis [4-8]. We present here a pediatric observation of osteosarcoma associated to retinoblastoma and osteosarcoma and we propose to discuss relation between these two malignant tumors.
Case Presentation
A 7-year-old boy presented to the consultation of one of the authors (MS) with a distal right
femur tumor (Figure 1 and 2). Previously, at two years of age, he was diagnosed to have unilateral retinoblastoma of the right eye. Enucleation of right eye followed by chemotherapy and radiotherapy
were performed. There were no other cases of retinoblastoma noted in the family medical history.
Osteosarcoma was confirmed by histological study after biopsy and treatment consisted of
neoadjuvant and adjuvant chemotherapy based on high dose of methotrexate as well as conservative
limb surgery. After trans-epiphyseal tumor resection, reconstruction was made by means of
autologous both fibulas (one fibula was vascularized and the other fibula was non-vascularized)
(Figure 3). Histological examination showed a very limited effect of chemotherapy and good
resection margins (Figure 4). Pseudarthrosis of the grafts occurred later and a cancellous graft was necessary in order for consolidation to be completed (Figure 5). At three years of follow-up, a pleural metastasis appeared causing death.
Figure 1
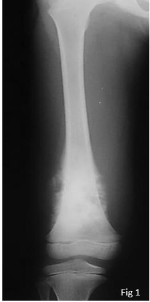
Figure 1
Disruption of cortical bone.
Figure 2
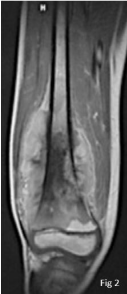
Figure 2
MRI of the femur, intraosseous aggressive process of the distal
portion of the femur, coming in contact with the cartilage with important
extension to the soft parts.
Discussion
Retinoblastoma may be hereditary or not [1]. The hereditary form of the disease involves a germ
cells mutation of the Rb gene and the not hereditary form involves a mutation in a cell in the retina.
It is well established that there is an increased risk of second cancers occurring after retinoblastoma
and the cumulative risk of SMN in retinoblastoma survivors is 32% [9]. This risk is more important
in the hereditary form and tends to decrease with age. Occurrence of SMN is significantly high in
patients with bilateral retinoblastoma [1,5,7,10].
A recent analysis of SMN in retinoblastoma survivors revealed that 76% of the second tumors
occurred in the head and neck region with a median age at diagnosis of 16 years [11]. Cumulative occurrence of SMN, fifty years after the diagnosis, is believed to be 36% in retinoblastoma survivors
[6].
A wide variety of soft tissue and bone second malignant tumors had been reported but the most common is for osteosarcoma [10,12]. Fujiwara et al. [13]
retrospectively reviewed a database of patients with retinoblastoma
and osteosarcoma occurring as a second malignancy between 1964
and 2010 at the National Cancer Center Hospital of Japan. Among 857
patients with retinoblastoma registered in the database, the authors
found 10 cases (1.1%) that developed secondarily osteosarcoma.
The nature of the association between retinoblastoma and second
osteosarcoma had had been studied extensively. The mutation of the
Rb (retinoblastoma) gene is strongly implicated in oncogenesis of
osteosarcoma. Patients with family history of retinoblastoma or those
having bilateral tumor, have an Rb1 germ-line defect and a higher risk
of developing an osteosarcoma than that of the general population
[5,6]. Indeed, this gene code for Retinoblastoma protein (pRb), a
regulatory protein of the cell cycle and its mutation causes the pRb
inactivation leading to the loss of osteoblasts cell cycle control and
promoting there by the initial tumor growth [7]. For some authors, the inactivation of pRb also leads to a loss of cell adhesion while promoting metastasis of osteosarcoma.
The risk of developing SMN in patients with retinoblastoma is
increased by radiotherapy, an additional treatment to surgery [1].
In a series of patients with bilateral retinoblastoma, Rotary et al.
[14] found a cumulative incidence of SMN of 35% for patients who
received radiation therapy and only 5.8% for patients who did not
receive radiotherapy. SMNs occur frequently on irradiated tissue and
rarely outside the field of irradiation [12,14].
For many authors, the prognosis of primary osteosarcomas is
believed to be better than second osteosarcomas that develop after
bilateral retinoblastoma. For successful treatment of osteosarcoma,
complete surgical resection is necessary [15-17]. However,
secondary osteosarcomas developing on irradiated tissue in the
craniofacial region are mostly inoperable, making their prognosis
very bad. Furthermore, the efficiency of chemotherapy in secondary
osteosarcomas is contested in retinoblastoma survivors [10]. This
might be due to the absence of the retinoblastoma protein in the human
sarcoma, causing the cells to be resistant to antimetabolites [16]. The
losses of heterozygosis at the Rb locus have been also implicated in
the poor prognosis of osteosarcomas [1]. However, in the series of Fujiwara et al. [13], four patients with tumors on an extremity were
treated by wide resection with neo adjuvant and adjuvant high-dose
methotrexate-based multi-agent chemotherapy and three of these
four patients (75%) were good responders to chemotherapy and
survived with no evidence of disease (median follow-up period, 17.3
years). The authors concluded that the clinical outcomes of second
osteosarcoma in an extremity occurring in retinoblastoma survivors
may be more favorable than those of conventional osteosarcoma.
In addition, the German-Austrian-Swiss cooperative study group
of osteosarcoma (COSS) had found that secondary osteosarcoma
treated by a combined approach could have a similar prognosis to
that of primary osteosarcoma [10].
Figure 3
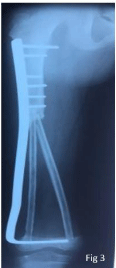
Figure 3
Trans-epiphysis tumor resection and reconstruction by the two
fibulas, one of which was vascularized.
Figure 4
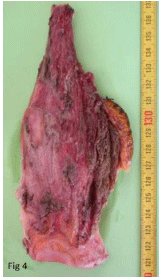
Figure 4
Tumor resection piece.
Figure 5
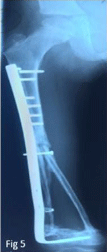
Figure 5
Radigraph of the right femur after addition of cancellous graft in
order for consolidation.
Conclusion
Faced with the risk of second sarcomas in patients treated for
retinoblastoma, regular monitoring and screening are required
for adequate management. Achieving a consensus on protocols of
chemotherapy seems to be necessary in order to standardize patient
care. A revision of irradiation techniques and radiation doses may
decrease the occurrence of SMN in the irradiated sites in patients
with retinoblastoma.
Finally, faced to the rarity of these cases, scientists have to develop
an international database for all these case reports in order to build
large series that will enable us to make more reliable guidelines and
conclusions.
References
- Abramson DH. Retinoblastoma in the 20th century: past success and future challenges. The Weisenfeld lecture. Invest Ophthalmol Vis Sci. 2005; 46: 2683-2691.
- Choi SY, Kim DH, Lee KM, Lee HJ, Kim MS, Lee TW, et al. Bilateral retinoblastoma: long-term follow-up results from a single institution. Korean J Pediatr. 2009; 52: 674-679.
- Armenian SH, Robison LL. Childhood cancer survivorship: an update on evolving paradigms for understanding pathogenesis and screening for therapy-related late effects. Curr Opin Pediatr. 2013; 25: 16-22.
- Blatt J, Olshan A, Gula MJ, Dickman PS, Zaranek B. Second malignancies in very-long-term survivors of childhood cancer. Am J Med. 1992; 93: 57- 60.
- Wong FL, Boice JD Jr, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. J Am Med Assoc. 1997; 278: 1262-1267.
- Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005; 23: 2272-2279.
- Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008; 100: 1771-1779.
- Hawkins MM, Kinnier Wilson LM, Burton HS, Michael HN Potok, Winter DL, Marsden HB, et al. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996; 88: 270-278.
- Tahasildar N, Goni V, Bhagwat K, Tripathy SK, Panda BB. Ewing's sarcoma as second malignancy following a short latency in unilateral retinoblastoma. J Orthop Traumatol. 2011; 12: 167-171.
- Bielack SS, Kempf-Bielack B, Heise U, Schwenzer D, Winkler K. Combined modality treatment for osteosarcoma occurring as a second malignant disease. J Clin Oncol. 1999; 17: 1164-1174.
- Venkatraman L, Goepel JR, Steele K, Dobbs SP, Lyness RW, Mac Cluggage WG. Soft tissue pelvic and urinary bladder leiomyosarcoma as second neoplasm following hereditary retinoblastoma. J Clin Pathol. 2003; 56: 233-236.
- Sagerman RH, Cassady JR, Tretter P, Ellsworth RM. Radiation induced neoplasia following external beam therapy for children with retinoblastoma. Am J Roentgenol Radium Ther Nucl Med. 1969; 105: 529- 535.
- Fujiwara T, Fujiwara M, Numoto K, Ogura K, Yoshida A, Yonemoto T, et al. Second primary osteosarcomas in patients with retinoblastoma. Jpn J Clin Oncol. 2015; 45: 1139-1145.
- Rotary JD, Mc Lean IW, Zimmerman LE. Incidence of second neoplasms in patients with bilateral retinoblastoma. Ophtalmology. 1988; 11: 1583- 1587.
- Agarwal G, Kochar HS, Julka PK, Bahadur S. Osteosarcoma as a second malignant disease in a case of bilateral retinoblastoma. Indian J Otolaryngol Head Neck Surg. 2011; 63: 115-117.
- Aerts I, Pacquement H, Doz F, Mosseri V, Desjardins L, Sastre X, et al. Outcome of second malignancies after retinoblastoma: a retrospective analysis of 25 patients treated at the Institut Curie. Eur J Cancer. 2004; 40: 1522-1529.
- Kay RM, Eckardtjj, Mira JM. Osteosarcoma and Ewing’s sarcoma in retinoblastoma patient. Clin orthop Rel Res. 1996; 323: 284-287.