Case Report
Does Autoimmunity have a Role in Myoclonic Astatic Epilepsy? A Case Report of Voltage Gated Potassium Channel Mediated Seizures
Deepa Sirsi*, Alison Dolce, Benjamin M Greenberg and Drew Thodeson
Department of Neurology, Neurotherapeutics and Pediatrics, UT Southwestern Medical Center, USA
*Corresponding author: Deepa Sirsi, Department of Neurology, Neurotherapeutics and Pediatrics, UT Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, Texas, USA
Published: 03 Nov, 2016
Cite this article as: Sirsi D, Dolce A, Greenberg BM,
Thodeson D. Does Autoimmunity have
a Role in Myoclonic Astatic Epilepsy?
A Case Report of Voltage Gated
Potassium Channel Mediated Seizures.
Ann Clin Case Rep. 2016; 1: 1178.
Abstract
Background: There is expanding knowledge about the phenotypic variability of patients with
voltage gated potassium channel complex (VGKC) antibody mediated neurologic disorders. The
phenotypes are diverse and involve disorders of the central and peripheral nervous systems. The
central nervous system manifestations described in the literature include limbic encephalitis, status
epilepticus, and acute encephalitis.
Patient Description: We report a 4.5 year-old boy who presented with intractable Myoclonic
Astatic Epilepsy (MAE) or Doose syndrome and positive VGKC antibodies in serum. Treatment
with steroids led to resolution of seizures and electrographic normalization.
Conclusion: This case widens the spectrum of etiologies for MAE to include autoimmunity, in
particular VGKC auto-antibodies and CNS inflammation, as a primary or contributing factor. There
is an evolving understanding of voltage gated potassium channel complex mediated autoimmunity
in children and the role of inflammation and autoimmunity in MAE and other intractable pediatric
epilepsy syndromes remains to be fully defined. A high index of suspicion is required for diagnosis
and appropriate management of antibody mediated epilepsy syndromes.
Keywords: VGKC antibody; Myoclonic astatic epilepsy; Doose syndrome; Autoimmune epilepsy; Generalized epilepsy
Introduction
Voltage gated potassium channel (VGKC) complexes of the central and peripheral nervous
system play an important role in synaptic transmission, conduction and repolarization [1]. Auto
antibodies to VGKCs are reported in a range of central and peripheral nervous system disorders
in adults and in children [2]. The proposed mechanism of pathogenicity is binding of antibodies
to transmembrane VGKCs which impair ion channel function leading to hyperexcitability. In the
central nervous system this leads to diffuse dysfunction, clinically manifesting as encephalopathy
and seizures [3]. There are a few case series and case reports about VGKC complex antibodies
and pediatric epilepsy. Previously reported patients had a common feature of encephalopathy but
otherwise had varying presentations including acute encephalitis, status epilepticus, febrile infection
related epilepsy syndrome (FIRES), and limbic encephalitis [2-8]. A larger case series by Dhamija
reported other manifestations such as movement disorders, gastro-intestinal dysmotility and small
fiber neuropathy [1].
Myoclonic-astatic epilepsy (MAE), or Doose syndrome, is classically considered an idiopathic
generalized epilepsy syndrome. Although several genetic and structural etiologies have been
purported, to our knowledge, there are no cases of MAE in the literature with an antibody mediated
mechanism [9,10]. Steroids have been reported as at least a partially effective treatment for MAE, although the mechanism of action and reason for lack of sustained response is unclear [9]. This case may provide some insights into the mechanism of action of steroids in MAE.
Clinically MAE is characterized by frequent myoclonic or atonic seizures with onset in early
childhood. In greater than fifty percent of patients, afebrile generalized tonic-clonic seizures may
appear first, prior to the onset of myoclonic-atonic seizures [11]. Generalized absence seizures
are variably present. Childhood development is normal prior to the onset of seizures. Initial
electroencephalogram (EEG) may also be normal with progression to generalized spike or polyspike and wave epileptiform activity and possible background slowing. Many cases are refractory to anti-seizure medications (ASM). We
present a child with characteristic features of MAE with positive
serumVGKC antibodies and excellent clinical and EEG response to
immunotherapy.
Case Presentation
A 4.5 year-old right-handed previously healthy and
developmentally normal boy presented to our emergency room (ER)
following new onset generalized tonic-clonic seizures. Intravenous
lorazepam and levetiracetam were administered for recurrent
seizures in the ER. He was admitted to the hospital where he had
frequent seizures characterized by brief (5-45 second) episodes of
staring with activity arrest, eye fluttering and clonic movements of the
face. He had no post-ictal confusion. An initial EEG was normal and
an MRI brain was unremarkable. Infectious workup, including urine,
blood and cerebrospinal fluid (CSF) cultures was negative. Despite initiation of maintenance levetiracetam (30 mg/kg/day) he continued
to have daily seizures but interictal mental status was normal. Three
days after presentation, a subsequent EEG showed continuous theta
slowing with frequent generalized spike and slow wave discharges
(Figure 1). After a four-day admission, he was discharged home on
levetiracetam (50 mg/kg/day).
The following day, he returned to the ER with recurrent,
frequent seizures now with bilateral clonic arm jerks and facial clonic
movements, involving the right more than left side. Valproic acid (15
mg/kg/day) was started and maintanance levetiracetam was increased
to 60 mg/kg/day. He continued to have repetitive seizures and
given the fulminant onset and refractory nature of seizures despite
rapidly titrating ASMs a pre-surgical evaluation was performed. EEG
demonstrated background slowing and generalized epileptiform
discharges. Seizures were clinically characterized by bilateral upper
extremity myoclonus, rapid eyelid fluttering, facial twitching and
subtle head drop. Ictal EEG showed 1-2 Hz generalized epileptiform discharges (Figure 2). Ictal SPECT was non-lateralized.
Further diagnostic workup included metabolic, genetic and
autoimmune studies. Workup included CSF amino acids, CSF
paraneoplastic panel, thyroid functions, liver and muscle enzymes,
serum amino acids, urine organic acids, acylcarnitine profile,
biotinidase, lactate, pyruvate, karyotype, chromosomal microarray
and serum paraneoplastic panel. A repeat MRI with MR spectroscopy
was also obtained and was unremarkable. There was no evidence
of central nervous system inflammation (CSF glucose 60 mg/dL,
CSF protein 17 mg/dL, acellular CSF, CSF oligoclonal bands and
neopterin were not obtained). At the time of discharge, it was noted
that he had subtle dysmetria and an ataxic gait which was attributed
to medication effect.
One month after discharge he remained seizure free on
levetiracetam (60 mg/kg/day) and valproic acid (15 mg/kg/day). Ticlike
behaviors with sudden vocalization and facial twitches were noted.
His mental status and development remained normal for age. He
continued to havesubtle dysmetria. His karyotype returned abnormal
showing a polymorphic centromeric variant on chromosome 4
(p12q12) of unclear significance. Serum paraneoplastic panel was
abnormal with positive VGKC antibodies (891 pmol/L; positive > 650
pmol/L). Western blot analysis for LGI1 and Caspr2 antibodies were
negative. CSF paraneoplastic panel was normal.
Given the unusual presentation without evidence of
encephalopathy and apparent seizure resolution, serum paraneoplastic
panel and EEG were repeated. CT imaging of the chest, abdomen and
pelvis was completed with no evidence of neoplasm. The follow up
video-EEG continued to demonstrate background slowing along with
generalized epileptiform abnormalities. In addition, the patient was
noted to have electroclinical seizures characterized by eye fluttering
and head drop with associated 5-10 second runs of generalized 1-2
Hz spike and slow wave discharges. These events were unnoticed by
the family.
Repeat serum paraneoplastic panel continued to show positive
VGKC antibodies (884 pmol/L), and western blot for LGI1 and Caspr2 remained negative. He was admitted for immunotherapy
and given a five-day course of intravenous methylprednisolone
(30 mg/kg/dose). A follow up 24-hour video-EEG after 5 days of
methyl prednisolone demonstrated no further seizures. In addition,
improvement in the EEG was noted with near normalization of the
background and only two brief runs of generalized epileptiform
discharges during a 24 hour EEG (Figure 3). He was discharged home
on the same ASMs with instructions to complete an oral prednisolone
taper over two months, starting at 1.2 mg/kg/day. Approximately
three weeks after completing steroid taper the family called to report
a break through seizure. However, this seizure was in the setting of
missed medications and with resumption of scheduled levetiracetam
and valproic acid he became seizure free again.
At 1 year follow up he remained seizure free. A repeat routine
EEG and a subsequent 4-hour EEG were normal. The previously
noted dysmetria and ataxia were no longer present and his parents
reported resolution of his tic like behaviors. Repeat VGKC antibodies
remained elevated and essentially unchanged at 762 pmol/L. The
patient was tapered off of valproic acid and levetiracetam and at 17
month follow up he remained seizure free off all ASMs.
Figure 1
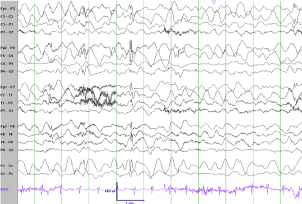
Figure 1
Interictal EEG 3 days after presentation. Bifrontal maximal generalized slowing of the background activity with generalized, bifrontal maximum spike and wave discharges.
Figure 2
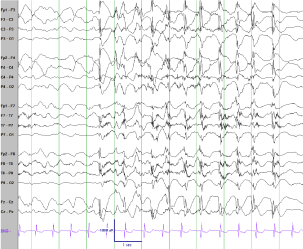
Figure 2
Ictal EEG. High amplitude generalized onset with evolving 1-2 Hz generalized spike and wave discharges correlated with activity arrest and eye fluttering.
Figure 3
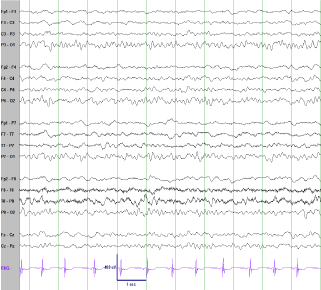
Figure 3
EEG 5 days after IV steroids. The EEG improved greatly, essentially normalizing with very infrequent generalized discharges (not shown here).
Discussion
There is increasing recognition of antibody mediated epilepsies,
specifically VGKC complex mediated autoimmune epilepsy. The
antibodies bind to proteins that are a part of the VGKC complex
such as leucine rich glioma-inactivated 1 protein (LGI1), contactin
associated protein 2 (Caspr2) or yet unidentified VGKC complex
proteins [1]. It is the most common of neuronal auto-antibodies
identified in adults evaluated for paraneoplastic autoimmunity in
some centers [1]. However, the incidence, and spectrum of VGKC
autoimmunity in children is yet to be established. The variable
clinical presentations may reflect different antigenic targets, age, comorbidities
and other factors that are yet to be determined [1].
Our patient’s clinical presentation is consistent with that of MAE,
or Doose syndrome. His normal development, constellation of seizure
types, and EEG findings support this diagnosis [9]. Children with MAE can have very small myoclonic movements that result in subtle
twitches and vocalizations. One might postulate that our patient’s
reported tic-like behaviors were in fact subtle myoclonic seizures.
Although MAE is a unique well-defined electro-clinical syndrome,
the underlying etiology is presumed to be genetic. Genetic mutations
including SCN1A, SCN1B, and GABRG2 have been identified in
a small number of families but single gene causes are rare. In our
patient, additional genetic testing (e.g. whole exome sequencing or an
epilepsy genetic panel) was not pursued as he had clinically improved
following steroid therapy and it was not felt to be cost-effective. MAE
has also been reported in structural epileptogenic conditions such as
Sturge-Weber syndrome and metabolic disorders such as cerebral
folate deficiency and the glucose transporter disorder, GLUT1
encephalopathy [12]. To our knowledge, there are no cases of VGKC
antibody mediated epilepsy presenting as a MAE, and we postulate
that this may be a diagnostic possibility with significant therapeutic
implications.
This case suggests that in children with MAE who have seizures
intractable to ASMs, paraneoplastic antibody testing may change
management and prognosis. Earlier case series of children have
reported encephalopathy as a prominent feature of VGKC antibody
autoimmunity and encephalitis with focal seizures [1,2]. Our patient
had no associated mental status change or psychiatric disturbance
and had generalized seizures possibly expanding the range of CNS
presentations of VGKC mediated disease. The noted subtle ataxia also
seems relevant as this improved following steroid treatment.
Although our patient’s serum yielded a positive result, there were
no VGKC antibodies present in his CSF. The explanation for this is
unclear but this may be due to limitations in testing methodologies.
A previous study of VGKC autoimmunity has reported reduced levels
(<1% to 10% of serum values) of VGKC antibodies in the CSF [13].
It has been hypothesized that the neuronal surface antibody in the
CSF may already be bound to the target antigen and therefore not
detectable in CSF [14]. Although elevated, the relatively low serum
antibody titers seen in our patient could be an epiphenomenon.
However, the dramatic clinical and electrographic response to IV
methylprednisolone followed by a regimen of oral prednisolone is an
argument for an autoimmune or inflammatory etiology even though
a true cause-effect relationship is not possible with this single case.
Steroid therapy has been reported as a treatment option in MAE and
some studies have reported seizure reduction but sustained seizure
freedom is rare in intractable MAE patients treated with steroids
[9,15]. A limitation of these studies was that they included multiple
different intractable pediatric epilepsy syndromes and they were
not specific to MAE. Anti-convulsive and anti-inflammatory effects
oral prednisolone and IV methylprednisolone are hypothesized as
mechanisms of action but the anti-epileptic mechanism of steroids
is not known [16].
Our case report raises the following questions. Is there a subset
of MAE patients where auto-immunity or CNS inflammation plays
an important role in epileptogenesis? Will identification of this
subset help guide the decision to treat with steroids? In our patient
steroid treatment allowed for reduction in AEDs from polytherapy
to complete withdrawal of all AEDs within one year after steroid
initiation.
With expanding understanding of VGKC antibodies, there is
also evidence to support that these antibodies may be a nonspecific
marker of CNS inflammation. In a recent publication, Dr. Sonderen and colleagues reported that only patients with VGKC-positive
antibodies along with LGI1 and/or Caspr2 positivity are clinically
relevant [17]. However, this was a small sample that only included 3
children. Further studies on the role of VGKC antibodies in children
with CNS disease are needed. In addition, larger systematic studies
investigating the role of autoimmunity and inflammation in MAE
syndrome, the characteristics of the MAE patient who will benefit
from steroid therapy and subsequently the optimal treatment regimen
with intravenous and oral steroids is needed.
Conclusion
This case highlights that VGKC complex antibodies can be associated with a diverse range of symptoms including new onset MAE that is intractable to ASMs. Causality of MAE by VGKC antibodies needs further investigation and cautious interpretation. Additional corroborating evidence of central nervous system involvement such as CSF pleocytosis, elevated protein, elevated neopterin, and presence of oligoclonal bands is helpful, but may be absent. There is a need for larger case series and multi-center trials investigating the etiology of unknown, presumed genetic, intractable childhood epilepsy syndromes. Recognition of autoimmune etiologies may in turn lead to appropriate and effective treatmentsand better prognosis for these children. Early identification could avoid continued trials of ASMs and exposure to their side effects. Prompt treatment with steroids or immune modulatory agents may lead to resolution of seizures and other manifestations. VGKC antibody testing may be considered in children who present with explosive onset of MAE that are refractory to first line ASMs even in the absence of clinical features of encephalitis or encephalopathy.
Acknowledgement
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award Number UL1TR001105. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- Dhamija R, Renaud DL, Pittock SJ, McKeon A, Lachance DH, Nickels KC, et al. Neuronal voltage-gated potassium channel complex autoimmunity in children. Pediatr Neurol. 2011; 44: 275-281.
- Lin JJ, Lin KL, Hsia SH, Wang HS, Chiu CH, et al. VGKC complex antibodies in pediatric severe acute encephalitis: A study and literature review. Brain Dev. 2013; 35: 630-635.
- Buckley C. Diseases Associated with Antibodies to Voltage-Gated Potassium Channels. Acnr. 2005; 5: 11-12.
- Kröll-Seger J, Bien CG, Huppertz HJ. Non-paraneoplastic limbic encephalitis associated with antibodies to potassium channels leading to bilateral hippocampal sclerosis in a pre-pubertal girl. Epileptic Disord. 2009; 11: 54-59.
- Haberlandt E, Bast T, Ebner A, Holthausen H, Kluger G, Kravljanac R, et al. Limbic encephalitis in children and adolescents. Arch Dis Child. 2011; 96: 186-191.
- Illingworth MA, Hanrahan D, Anderson CE, et al. Elevated VGKCcomplex antibodies in a boy with fever-induced refractory epileptic encephalopathy in school-age children (FIRES). Dev Med Child Neurol. 2011; 53: 1053-1057.
- Suleiman J, Brenner T, Gill D. VGKC antibodies in pediatric encephalitis presenting with status epilepticus. Neurology. 2011; 76: 1252-1255.
- Iyer A, McTague A, Curran A, Inbasagaran A, Vincent A, Kneen R, et al. VGKC-complex antibody mediated encephalitis presenting with psychiatric features and neuroleptic malignant syndrome - further expanding the phenotype. Dev Med Child Neurol. 2012; 54: 575-576.
- Kelley SA, Kossoff EH. Doose syndrome (myoclonic-astatic epilepsy): 40 years of progress. Dev Med Child Neurol. 2010; 52: 988-993.
- Oguni H. Symptomatic epilepsies imitating idiopathic generalized epilepsies. Epilepsia. 2005; 46: 84-90.
- Kilaru A, Bergqvist AG. Current treatment of myoclonic astatic epilepsy: clinical experience at the Children’s Hospital of Philadelphia. Epilepsia. 2007; 48: 1703-1707.
- Tang S, Pal DK. Dissecting the genetic basis of myoclonic-astatic epilepsy. Epilepsia. 2012; 53: 1303-1313.
- Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: A potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004; 127: 701-712.
- Sinmaz N, Amatoury M, Merheb V, Ramanathan S, Dale RC, Brilot F. Autoantibodies in movement and psychiatric disorders: Updated concepts in detection methods, pathogenicity, and CNS entry. Ann N Y Acad Sci. 2015; 1351: 22-38.
- You SJ, Jung DE, Kim HD, Lee HS, Kang HC. Efficacy and prognosis of short course of prednisolone therapy for pediatric epilepsy. Eur J Paediatr Neurol. 2008; 12: 314-320.
- Bakker D, Coriene E, Neuteboom RF. Effectiveness of hybrid corticosteroid treatment regimen on refractory childhood seizures and a review of other corticosteroid treatments. Eur J Paediatr Neurol. 2015; 19: 553-560.
- van Sonderen A, Schreurs MW, de Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES, et al. The relevance of VGKC positivity in absence of LGI1 and Caspr2 antibodies. Neurology. 2016; 86: 1692-1699.