Research Article
Fundus Albipunctatus: A Novel Mutation
Fadwa Al Adel1*, Irma Lopez2, Ayesha Khan2, Robert Koenekoop2, Julie Racine3 and Ahmed Basheikh4
1Department of Ophthalmology, Princess Nourah bint Abdulrahman University, Saudi Arabia
2Department of Ophthalmology, McGill University, Canada
3Department of Ophthalmology, Eye Clinic, Nationwide Children's Hospital, United States
4Department of Ophthalmology, King Abdulaziz University, Saudi Arabia
*Corresponding author: Fadwa Al Adel, Department of Ophthalmology, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia
Published: 15 Sep, 2016
Cite this article as: Adel FA, Lopez I, Khan A, Koenekoop
R, Racine J, Basheikh A. Fundus
Albipunctatus: A Novel Mutation. Ann
Clin Case Rep. 2016; 1: 1135.
Abstract
Background: The aim of this paper is to describe a patient with fundus albipunctatus from Africa
who is developing a cone dystrophy with a new mutation in RDH5.
Methods: A 24-year-old female from Burundi presented with difficulty to adapt to darkness after
being in the light since the age of 9 years old. The clinical history, visual field, electroretinogram
(ERG), dark adaptation test, spectral-domain optical coherence tomography (SD-OCT); fundus
autofluorescence (FAF) and polymerase chain reaction (PCR) plus DNA analysis (by Sanger
sequencing) were performed.
Results: Fundus examination revealed scattered, homogenous, yellow-white dots throughout both
fundi. OCT showed extrafoveal numerous well-demarcated homogenous dome-shaped lesions
originating in the RPE. FAF showed a lack of autofluorescence (AF). Electroretinogram of the cone
and the mixed rod-cone system had a normal morphology but with a significant decrease in the a and
b wave amplitudes and a delayed peak time. The rod-mediated ERG was non-detectable. However,
after a prolonged (3-hour) period of dark adaptation, the rod-mediated ERG was detectable and
the b-wave reached 48% of normal value. The DNA analysis and sequencing of RDH5 revealed a
homozygous c. 524-526delACT mutation, leading to a protein change of p.Tyr175del, which has
not been reported before. The cone ERG results, strongly suggest that she is developing a cone
dystrophy, despite the fact that she is asymptomatic and still maintains 20/20 vision and normal
color discrimination.
Conclusion: We identified a new deletion in RDH5 responsible for fundus albipunctatus with a
progressive cone dystrophy; c. 524-526delACT in a patient from Burundi.
Keywords: Fundus albipunctatus; RDH5; Cone dystrophy; 11-cis retinol dehydrogenase; Dark adaptation; Spectral-domain optical coherence tomography (SD-OCT); Fundus autofluorescence
Introduction
Fundus albipunctatus (FA) is an autosomal recessive congenital night blinding disorder
characterized by the presence of retinal white dots caused by mutations in the RDH5 gene [1,2], encoding for the retinol dehydrogenase (RDH). RDH acts in the retinoid cycle to replenish 11 cis
retinal and is responsible, for the oxidation of 11-cis-retinol to 11-cis-retinal [3,4], the last step of the vitamin A cycle.
In the RPE cell somata, 11-cis retinol dehydrogenase (11-cis RDH) is an abundantly expressed
transmembrane protein [1,5]. The enzyme catalyzes the final step in the visual cycle, the biosynthesis of 11-cis retinal before it binds to opsin in rods and to the cone opsin in cones, to form rhodopsin
and the cone opsins [6].
A clinically distinct disorder, retinitis punctata albescens (RPA) is also characterized by
nyctalopia, reduced visual acuity, multiple round white deposits in the retina, but has progressive
attenuation of retinal arterioles, and non-detectable or severely reduced ERG recordings [7]. The first distinction between FA and RPA was made by Lauber (1910) [8]. RPA is associated mostly with mutations in the RLBP1 gene and occasionally in RHO, RDS, and RDH5 [9].
Fundus albipunctatus has a variable regeneration of dark adaptation and full-field electroretinography (ffERG) responses after prolonged dark adaptation, which helps in differentiating it from other disorders of the visual cycle.
Currently 22 RDH5 mutations are known [10]. In this article, we
present clinical and molecular genetic data in a 24-year-old African
woman with fundus albipunctatus, with a new deletion in RDH5, the
c. 524-526delACT mutation.
Figure 1
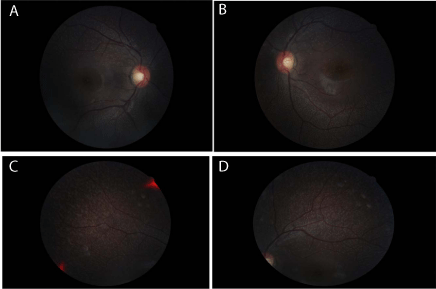
Figure 1
Fundus photograph of the patient: (a, b) Posterior pole; (c, d) Mid
periphery.
Figure 2
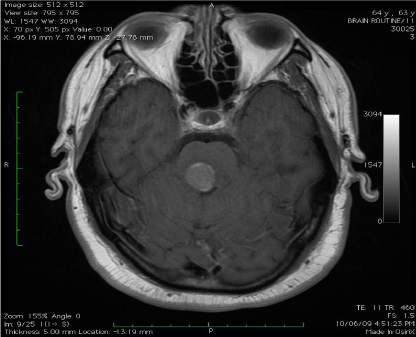
Figure 2
SD-OCT of the patient: (a) Posterior pole; (b) Mid periphery.
Materials and Methods
The patient provided written informed consent on behalf of
herself in accordance with the Declaration of Helsinki. Ethical and
scientific approvals for our studies have been approved by McGill
University Health Centre.
Clinical exam included visual acuity assessment with Snellen
chart, color vision with Ishihara plates, dilated fundus exam, color
photography, SD-OCT, FAF were obtained with a confocal scanning
laser ophthalmoscope, Goldmann visual field, ERG, dark adaptation
test, blood extraction for genetic analysis.
ERGs were done in accordance with International Society for
Clinical Electrophysiology of Vision (ISCEV). Short flash (20 μs;
white xenon light) cone- (background: 30 cd.m-2; flash intensity:
0.69 log cd.sec.m-2; ISI: 1.5 sec), rod- (dark adaptation: 15 minutes
and 3 hours; flash intensity: -2.3 log cd.sec.m-2; ISI: 10 sec) and rod/
cone- (dark adaptation: 15 minutes and 3 hours; flash intensity: 0.69
log cd.sec.m-2; ISI: 10 sec) mediated electroretinograms (ERGs) were
recorded using the Espion E2 system (Espion E2 system stimulator,
Diagnosis LLC, Lowell, MA, USA). The recording bandwidths were
set from 1.25 to 300 hertz. The a-wave amplitude was measured from the pre-stimulus
baseline to the first negative trough of the ERG, whereas the b wave
amplitude was measured form the trough of the a-wave to the most
positive peak of the ERG. For rod-mediated ERGs, where there is no
a-wave, the b-wave amplitude was measured from the pre-stimulus
baseline to the most positive peak of the ERG. Peak times of both the
a- and b-waves were calculated from the flash onset to the respective
wave peak.
DNA was extracted from whole blood by using FlexiGene kit
according to the manufacturer’s protocol. NanoDrop was used to
check DNA’s quantity and quality. Primer3 software was used to
design the RDH5 primers (http://frodo.wi.mit.edu/primer3/). The
HotStarTaq Master Mix kit from QIAGEN was used to perform
the PCRs following the manufacturer’s protocol. The purity and
specificity of the fragment was verified by agarose gel electrophoresis.
Then the fragment was sent for sequencing by Sanger sequencing
using Applied Biosystems 3730xl DNA Analyzer technology. The
DNA Sequencing Software Sequencher from Gene Codes was used
to analyze the results.
Results
This 24 year old black female from Burundi, presented to the
hereditary retinal degeneration clinic, McGill Ocular Genetics
Laboratory (MOGL) at the Montreal Children’s Hospital, with a
complaint of difficulty adapting to the dark after being in the light,
since she was 9 years old. She was also complaining of nyctalopia
which was improving with her age. Her past medical history was
insignificant and she was not on any medications. She came from
a non-consanguineous family, and had a sister who was living in
Burundi with the similar complaint.
The patient underwent a complete ocular exam. Her visual acuity
was 20/20 in both eyes. Her color vision was normal in both eyes.
There was no afferent pupillary defect. The intraocular pressure and
the anterior segments were normal for both eyes. Fundus examination
revealed scattered, homogenous, yellow-white dots throughout both
fundi, relatively sparing the macula (Figure 1).
The optic nerve had a normal color with a normal cup-to-disc
ratio. The retinal vessels were of normal caliber size.
A Spectral-Domain Optical Coherence Tomography (SDOCT)
exam was performed, and showed the presence of extrafoveal
numerous well-demarcated homogenous dome-shaped lesions,
extending from the retinal pigment epithelium (RPE) level into the
inner/outer segment junction of the photoreceptors (Figure 2).
Images for fundus autofluorescence (FAF), showed some
increased autofluorescence corresponding to the white dot lesions
(Figure 3).
The patient underwent a Goldmann manual perimetry, which
was normal in both eyes.
The cone-mediated electroretinograms recorded from the patient
revealed significantly decreased a- and b-wave amplitudes. More
specifically, the a- and b-waves were decreased by 60% and 65%
respectively compared to normal values. Their peak times were also
delayed by approximately 3-4 ms. While the rod-mediated ERG
response did not demonstrate a b-wave after 15 minutes of dark
adaptation, a small b-wave could be recorded after a 3-hour period
of dark adaptation. The amplitude of the rod-mediated b-wave after prolonged dark adaptation was 48% of normal values with
delayed peak times (approximately 10 ms). Finally, the rod/conemediated
ERG recorded after 15 minutes of dark adaptation showed
significantly decreased a- and b-wave amplitudes, 28% and 15% of
normal values respectively, with abnormal peak times. However,
after a 3-hour period of dark adaptation the amplitude of the a- and
b-waves increased to reach values that were 85% (a-wave) and 69%
(b-wave) of normal values. Peak times remained abnormal even with
prolonged dark adaptation (Figure 4).
PCR, DNA analysis and sequencing of the RDH5 gene revealed
a new deletion mutation in the RDH5, c. 524-526delACT mutation,
which has not been previously reported (Figure 5).
Figure 3
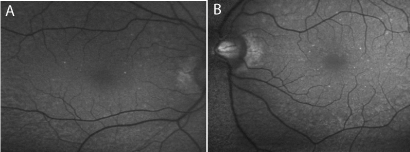
Figure 3
Fundus auto-fluorescence of the patient: (a) Right eye, (b) Left eye.
Figure 4
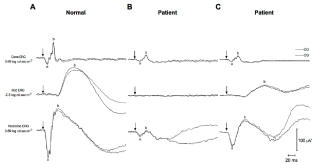
Figure 4
Right (OD; full line) and left (OS; dash line) eye short flash cone-,
rod- and rod/cone-mediated electroretinograms (ERGs) recorded from a (a)
normal subject and from a patient affected with fundus albipunctatus (Patient)
recorded either after a (b) 15-minute or a (c) 3-hour period of dark adaptation.
(a) a-wave, (b) b-wave, (arrows) flash onset. Horizontal calibration: 20 mV.
Vertical calibration: 100 μV.
Figure 5
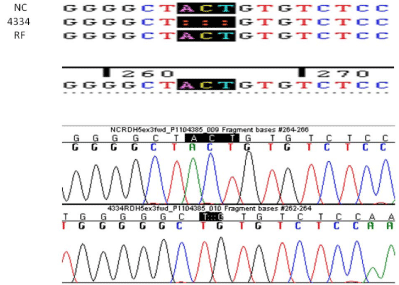
Figure 5
DNA analysis and sequencing of the RDH5 gene.
Discussion
The SD-OCT exam in our FA patient showed numerous outer
retinal lesions at the level of the retinal pigment epithelium, which
extended into the overlying inner segment/outer segment (IS/OS)
junction of the photoreceptors as well as the outer nuclear layer. This
finding is similar to a recent finding reported by Genead [11].
The cone-mediated electroretinograms from our patient was
significantly reduced in both a- and b- wave amplitudes which
indicates a cone dystrophy, and implies that patient will get worse and
that the disease is not a stationary disease. To prove this hypothesis, it
is necessary to observe patients with FA for a long period.
To complicate the differential diagnostic process in the clinical
setting, improvement of the dark-adapted ERG responses following
prolonged dark adaptation has been reported also in patients with RPA [12]. However, in RPA patients with RLBP1 mutations, recovery
of ERG responses to the normal range has been observed only after
20 or more hours of dark adaptation [12] and apparently not after a
shorter (10 h) adaptation time [13]. No recovery after 2 h and only
a modest recovery after 17 h has also been recently reported in a
RLBP1 -negative RPA patient [7]. Our patient had an increase in the
amplitude of the rod/cone-mediated ERG by 85% (a-wave) and 69%
(b-wave) of normal values after a 3-hour period of dark adaptation.
The cone ERG results, strongly suggest that she is developing a
cone dystrophy, despite the fact that she is asymptomatic and still
maintains 20/20 vision and normal color discrimination.
Our molecular genetic analysis demonstrates a new deletion
mutation in RDH5, a homozygous 3bp deletion c. 524-526delACT.
Some patients with FA and RDH5 mutations are stable, while
others develop cone dystrophy. The genetic changes found in RDH5
do not appear to predict which will progress and which will be stable.
Conclusion
In conclusion, we present a case of a young patient from Burundi from a non-consanguineous family with fundus albipunctatus who is developing a progressive cone dystrophy with a new deletion mutation in the RDH5 gene. We discovered that despite her 20/20 vision, she has developed a subclinical cone dystrophy that will progress over time.
Acknowledgments
We thank the family involved. Financial support for the study comes from Foundation Fighting Blindness Canada (FFB), CIHR, FRSQ and NIH (to RKK).
References
- Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, Dryja TP. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999; 22: 188-191.
- Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000; 130: 547-563.
- Mata NL, Radu RA, Clemmons RC, Travis GH. Isomerization and oxidation of vitamin a in cone-dominant retinas: a novel pathway for visual-pigment regeneration in daylight. Neuron. 2002; 36: 69-80.
- Kim TS, Maeda A, Maeda T, Heinlein C, Kedishvili N, Palczewski K et al. Delayed dark adaptation in 11 cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J Biol Chem. 2005; 280: 8694– 8704.
- Huang J, Possin DE, Saari JC. Localizations of visual cycle components in retinal pigment epithelium. Mol Vis. 2009; 15: 223–234.
- Schatz P, Preising M, Lorenz B, Sander B, Larsen M, Rosenberg T. Fundus Albipunctatus Associated with Compound Heterozygous Mutations in RPE65. Ophthalmology. 2011; 118: 888-894.
- Fishman GA, Roberts MF, Derlacki DJ, Grimsby JL, Yamamoto H, Sharon D et al. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch Ophthalmol. 2004; 122: 70-75.
- Lauber H. Die sogenannte Retinitis punctate albescences. Klin Monatsbl Augenheilkd. 1910; 48: 133–148.
- Humbert G, Delettre C, Sénéchal A, Cécile Bazalgette, Abdelhamid Barakat, Christian Bazalgette et al. Homozygous deletion related to Alu repeats in RLBP1 causes retinitis punctata albescens. Invest Ophthalmol Vis Sci. 2006; 47: 4719–4724.
- http://www.hgmd.cf.ac.uk/ac/index.php
- Genead MA, Fishman GA, Lindeman M. Spectral-domain optical coherence tomography and fundus autofluorescence characteristics in patients with fundus albipunctatus and retinitis punctata albescens. Ophthalmic Genet. 2010; 31: 66-72.
- Granse L, Abrahamson M, Ponjavic V, Andreasson S. Electrophysiological findings in two young patients with Bothnia dystrophy and a mutation in the RLBP1 gene. Ophthalmic Genet. 2001; 22: 97–105.
- Burstedt MS, Sandgren O, Golovleva I, Wachtmeister L. Retinal function in Bothnia dystrophy. An electrophysiological study Vision Res. 2003; 43: 2559–2571.