Case Report
Amyopathic Dermatomyositis: A Challenging Diagnosis
Chora I*, Ourique C, Silva S and Vaz-Marques P
Department of Internal Medicine, Centro Hospitalar São João, Portugal
*Corresponding author: Inês Chora, Department of Internal Medicine, Centro Hospitalar São João, Al. Hernâni Monteiro, 4200-319 Porto, Portugal
Published: 17 Aug, 2016
Cite this article as: Chora I, Ourique C, Silva S,
Vaz-Marques P. Amyopathic
Dermatomyositis: A Challenging
Diagnosis. Ann Clin Case Rep. 2016;
1: 1083.
Abstract
The authors report the case of a 68-year-old woman, first admitted for hypoxemic respiratory insufficiency and constitutive symptoms. Mild asymmetric arthritis, increased erythrocyte sedimentation rate and positive anti-nuclear antibodies raised the suspicion of a connective tissue disease and corticosteroids were started, with initial clinical improvement. Respiratory study revealed mild fibrosis on CT-scan and an obstructive ventilatory defect, interpreted as tuberculosis sequelae. Weeks later, the patient developed proximal muscle weakness and solid dysphagia. Normal muscle enzymes, the exclusion of a demyelinating disease, electromyography and muscle biopsy suggested a non-inflammatory glucocorticoid-induced myopathy, with clinical improvement following prednisone-tapering. A heliotrope rash appeared three months later. The diagnosis of dermatomyositis, with interstitial lung disease and of amyopathic subtype was then clear. Corticosteroids and immunosuppressants were started, with initial clinical worsening and readmission for large pneumomediastinum, but subsequent improvement. Cancer screening was negative.
Keywords: Amyopathic dermatomyositis; Interstitial lung disease; Glucocorticoid-induced myopathy
Case Presentation
We present the case of a 68-year-old Caucasian woman admitted to the hospital for an acute
hypoxemic respiratory failure and constitutive symptoms. She had a relevant past medical history
of treated pulmonary tuberculosis (PT) and was not taking any medication. Her brother suffered
from psoriasis.
The patient presented erythematous scaly cutaneous lesions on elbows, bilateral edema of wrists,
arthritis of distal and proximal inter-phalangeal joints of right hand’s second finger, bilateral fine
crackles on lower lung fields and no other relevant findings. Pulmonary embolism was excluded and
pulmonary computed tomography (CT) showed sub-pleural linear opacities at left superior lobe,
fibrotic striae at pulmonary bases, cystic bronchiectasis of right pulmonary base and centrilobular
micronodules at inferior pulmonary lobes (Figure 1).
Laboratory studies revealed mild lymphopenia, negative C-reactive protein, elevated
erythrocyte sedimentation rate (ESR) (89 mm/hour) and positive anti-nuclear antibodies (ANA)
(1/1000, speckled pattern). Muscle enzymes (creatine kinase, lactate dehydrogenase, aldolase and
myoglobin) and further immunologic results were normal. Microbiological exams were all negative.
Echocardiography and bronchoscopy were normal and both bronchoalveolar and bronchial lavage
fluids were negative for malignant cells and microbiologic testing. No antibiotics were prescribed.
Pulmonary function tests (PFT) showed a mild obstructive ventilatory defect which, again with the
imaging changes, was attributed to PT sequelae.
Constitutive symptoms, joint involvement and analytic results raised the suspicion of a
connective tissue disease (CTD), still undifferentiated, and the patient was started on oral prednisone
(40mg daily) with clinical improvement. Two weeks after being discharged, she was observed
at the outpatient clinic. She complained of fatigue and developed a severe and predominantly
proximal muscle weakness, as well as solid dysphagia. Serum levels of muscle enzymes remained
normal. Electromyography was normal, cerebral magnetic resonance imaging was not compatible
with demyelinating disease and deltoid muscle biopsy presented non-inflammatory changes and
suggested a glucocorticoid-induced myopathy (GIM). Prednisone was tapered and subsequent
clinical improvement on muscle strength supported the diagnosis of GIM; the patient continued
tapering prednisone.
Three months later, a heliotrope rash appeared (Figure 2) and the diagnosis of dermatomyositis (DM) with interstitial lung disease (ILD) was established, as well as its amyopathic subtype. The patient was started on oral prednisone (1mg/kg/day) and oral azathioprine (1.5mg/kg/day). Cancer screening was negative.
One month after the diagnosis of amyopathic DM (ADM), the patient was readmitted for syncope. Pulmonary CT scan showed a marked worsening of structural changes and a spontaneous pneumomediastinum (Figure 3). Azathioprine was switched to cyclophosphamide (CPA) and a first intravenous pulse (0.6g/m2) was given. A slight decrease of the pneumomediastinum was documented by additional CT scans. The patient remained clinically stable and was discharged; a next IV pulse of CPA was programmed within one month. Two weeks after the second pulse of CPA, the patient was readmitted for a larger spontaneous pneumomediastinum, with cervical subcutaneous emphysema, that clinically improved after supplemental oxygen and absolute resting.
After 12 monthly pulses of CPA, the patient was able to perform her daily life activities without supplemental oxygen. The control CT scan showed a similar degree of pulmonary fibrosis, no worsening of structural changes related to ILD and no pneumomediastinum. The patient performed a six-minute walk test without significant desaturation.
Figure 1
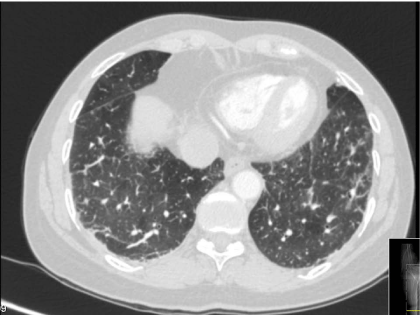
Figure 1
Pulmonary CT-scan on first admission, showing sub-pleural opacities, fibrotic striae and centrilobular micronodules.
Figure 2
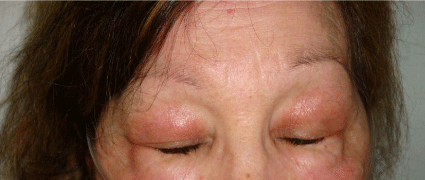
Figure 2
Heliotrope rash.
Figure 3
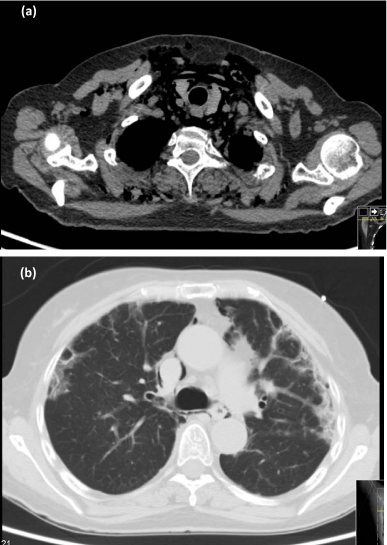
Figure 3
Thoracic high resolution CT-scan on second admission, showing pneumomediastinum(a) and sub-pleural fibrosis (b).
Discussion
ADM is a rare but well-recognized subset of DM (approximately 20% of cases) [1]. It is characterized by biopsy-confirmed cutaneous manifestations of classic DM occurring for six months or longer, with no clinical evidence of proximal muscle weakness and no serum muscle enzyme or other muscle testing abnormalities [2].
This case represents a diagnostic challenge. In the beginning, the constitutive symptoms, joint involvement, positive ANA and increased ESR raised the hypothesis of CTD, although the patient did not fulfill any classification criteria for a specific disease. In this context, and after excluding active infection, glucocorticoids were prescribed. Pulmonary disease was first interpreted not as ILD but as a consequence of PT sequelae, after excluding pulmonary infection and according to patient’s previous history and PFT.
The patient developed muscle weakness after beginning oral prednisone. This fact, again with normal muscle enzymes, normal electromyography, histological findings and clinical improvement few weeks after prednisone tapering, allowed the diagnosis of GIM. GIM is a frequent but often underestimated adverse effect of steroid treatment, being more common in elderly patients and appearing few weeks to several months after corticotherapy introduction [3]. Corticoid doses correlate positively with the likelihood of developing myopathy and with a faster onset of weakness. GIM eventually resolves if steroid therapy is discontinued.
In this patient, and after steroid discontinuation, the appearance of a single clinical sign - the heliotrope rash - permitted the whole reinterpretation of clinical data. This case reinforces that clinical suspicion is extremely important and differential diagnosis must be actively explored, even if rare, especially if they can course with rapidly progressive clinical deterioration; it also illustrates that “clinical picture” can change within time, so patients should be closely followed.
ADM diagnosis was established: the initial cutaneous lesions on elbows (Gottron’s sign) were present for more than six months, with no clinical evidence of inflammatory myositis as described above (classical DM myopathy improves with steroids) [4]. We have not found any report of concomitant ADM and GIM on literature search.
ILD has been reported in 13% of ADM patients, being frequently rapidly progressive and potentially fatal and requiring intensive management [1,5]. NSIP is most commonly reported [5]. Anti-CADM-140 antibody might be a risk factor for the development of ILD in ADM patients [1]. ILD can precede the diagnosis (18-20% of patients), appear simultaneously or develop later [5]. In this patient, ILD was present from the onset of skin lesions but was misinterpreted. Early treatment with high-dose corticosteroids and additional immunosuppressive agents may result in improved outcomes for some patients. Several case series attest the benefit of CPA, being reserved for severe or refractory cases given its serious toxicities [4,5].
Managing this patient’s disease course was challenging. At the time of ADM diagnosis, she started high-dose corticosteroids plus azathioprine but the delay regarding her first admission may be associated with a worse outcome. ADM-related ILD had a rapid progression and was refractory to first-line immune suppression, with markedly worsening of structural pulmonary changes and complicating with severe pneumomediastinum, which relapsed even after the first CPA course but finally improved. CPA is typically administered for six months before switching to another immunosuppressive agent; however, in this patient, it was decided to maintain CPA pulses for one year, considering the severity of the disease.
Spontaneous pneumomediastinum is a rare and severe complication of ILD in myositis, occurring more frequently in ADM [5]. Poor survival was associated with absence of muscle weakness and severe pulmonary involvement before the onset of pneumomediastinum [6].
ADM patients may be at increased risk of developing malignancy, though its rate in comparison to classic DM needs further studies [1]. Although the diagnosis of ADM in elderly patients increases the suspicion of an underlying malignancy, this patient’s cancer screening was negative. Close surveillance should continue for at least three to five years after the diagnosis of DM, as malignancy risk decreases to close to baseline after this period; then, age-appropriate cancer screening is recommended [1].
Learning Points
Amyopathic dermatomyositis is frequently associated with a rapidly progressive and severe interstitial lung disease (ILD) that may precede its diagnosis. Initial aggressive treatment should be considered if pulmonary complications develop.
• A high index of suspicion for the presence of ILD in the setting of dermatomyositis (DM) is mandatory; conversely, in patients with seemingly idiopathic ILD, and even in the absence of typical muscle involvement, DM should be ruled out.
• Glucocorticoid-induced myopathy must be considered in suitable clinical scenarios, as its diagnosis influences therapeutic choices.
References
- Ghazi E, Sontheimer RD, Werth VP. The importance of including amyopathic dermatomyositis in the idiopathic inflammatory myositis spectrum. Clin Exp Rheumatol. 2013; 31: 128-134.
- Gerami P, Schope JM, Mcdonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006; 54: 597–613.
- Perrot S, Le Jeunne C. Steroid-induced myopathy. Presse Med. 2012; 41: 422-426.
- Vincze M, Danko K. Idiopathic inflammatory myopathies. Best Pract Res Clin Rheum. 2012; 26: 25-45.
- Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013; 143: 814-824.
- Le Goff B, Cherin P, Cantagrel A, Gayraud M, Hachulla E, Laborde F, et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum. 2009; 61: 108-118.