Case Report
A Case Report: Non-Obstructive Azoospermia and Normal Phenotype in a Thirty-Eight Year Old Male with a Rare Klinefelter’s Syndrome Genotype
Mia Letterie1, Janet L Kennedy2* and R Dale McClure2
1Whitman College, USA
2Department of Reproductive Medicine, Seattle Reproductive Medicine, USA
*Corresponding author: Janet L Kennedy, Department of Reproductive Medicine, Seattle Reproductive Medicine, 1505 Westlake Ave, Suite 400, Seattle, WA 98109, USA
Published: 21 Jul, 2018
Cite this article as: Letterie M, Kennedy JL, McClure
RD. A Case Report: Non-Obstructive
Azoospermia and Normal Phenotype
in a Thirty-Eight Year Old Male with a
Rare Klinefelter’s Syndrome Genotype.
Ann Clin Case Rep. 2018; 3: 1537.
Abstract
Background: Klinefelter’s syndrome (KS) is the most common genetic cause of infertility in males.
KS is characterized by non-obstructive azoospermia (NOA) most often accompanied by physical
signs including small testes, tall stature, and gynecomastia. The most common karyotype is 47,
XXY. We describe a patient with a rare supernumerary isochromosome of X and relatively normal
phenotype. We provide evidence regarding the approach to treatment of infertility in these patients.
Case Summary: We describe an infertile male who presented only with non-obstructive
azoospermia and small testes with no other phenotypic manifestations of Klinefelter’s Syndrome.
His karyotype, 47, X,i(Xq),Y, includes a rare isochromosome of X. Given the absense of physical
evidence of hypogonadism, the patient and his wife held some hope that sperm would be found on
testicular biopsy. No sperm were identifiable after bilateral microsurgical testicular sperm extraction
(mTESE). Alternative treatment options were suggested and the couple eventually conceived via
insemination with donor sperm.
Conclusion: Given the rarity of this presentation, this information may provide direction in future
treatment by confirming that surgical sperm retrieval remains futile even in KS patients with a
supernumerary isochromosome X despite a relatively normal phenotype. Treating directly with
therapeutic donor insemination has a far higher pregnancy rate and avoids an unnecessary and
expensive treatment attempt.
Keywords: Klinefelter’s syndrome; Non-obstructive azoospermia; Isochromosome;
Microsurgical testicular sperm extraction
Abbreviations
KS: Klinefelter’s Syndrome; NOA: Non-Obstructive Azospermia; TESE: Testicular/Epididymal Sperm Extraction; mTESE: Microsurgical Testicular Sperm Extraction
Introduction
Klinefelter’s syndrome is the most common form of male hypogonadism and the most common genetic cause of infertility in males (4% of newborn males) [1]. KS is characterized by non-obstructive azoospermia (NOA) most often accompanied by a variety of clinical signs. The three most common physical attributes that suggest the diagnosis include: small testes, tall stature and gynecomastia [2]. The most common karyotype is 47, XXY accounting for 80% to 90% of patients with KS [3]. Rare variants of this karyotype have been described with varying clinical features. We describe an infertile male who presented only with NOA and small testes with no other phenotypic manifestations of Klinefelter’s Syndrome. His karyotype, 47 X, i(Xq), Y, includes a rare isochromosome of X. Given the absence of physical evidence of hypogonadism, the patient and his wife held some hope that sperm would be found on testicular biopsy. No sperm were identifiable after bilateral microsurgical testicular sperm extraction (TESE). Alternative treatment options were suggested and the couple eventually conceived via insemination with donor sperm.
Case Presentation
The 38 year old patient presented for evaluation of azoospermia after a semen analysis was
performed as part of an infertility evaluation. He and his 29 year old spouse had been attempting pregnancy for approximately 36 months. Additional inquiry after
discovery of this finding revealed a history of delayed (age 16) but
otherwise normal puberty and a clinical exam that demonstrated
bilateral small testicular volume. No further diagnosis was given and
the subject was referred to our clinic.
On physical examination by one of the authors, the patient
was 71 inches tall, a normal stature for his family, had no signs of
gynecomastia and exhibited normal virilization. There was no deficit
in cognition. Ultrasound examination with testicular measurements
indicated dimensions of 2.0 cm × 2.0 cm on the left and 2.0 cm ×
1.0 cm on the right. No hyperechoic foci were noted by ultrasound
[4]. Laboratory evaluation indicated a normal total testosterone level
(415 ng %), elevated serum FSH (36.8 mIU/mL) and LH (25.3 mIU/
mL) concentrations, and a normal TSH (1.3 mcIU/ml) level. His
karyotype was found to be 47, X,i(Xq),Y (Figure 1).
The couple initially refused treatment with donor sperm and
elected to proceed with testicular sperm aspiration. A bilateral
scrotal exploration and attempted microsurgical sperm retrieval
was performed by one of the authors. On the left side, dissection
was carried down to the testis itself. The tunica vaginalis was opened
and the testis was bivalved horizontally. Microscopic examination of
ten biopsies revealed no sperm present. On the right side a similar
procedure was performed. Again, more than 10 biopsies were taken
and no sperm were identified.
After the testicular exploration failed to identify sperm, the
couple decided to proceed with donor sperm insemination, achieved
a pregnancy and term delivery.
Figure 1
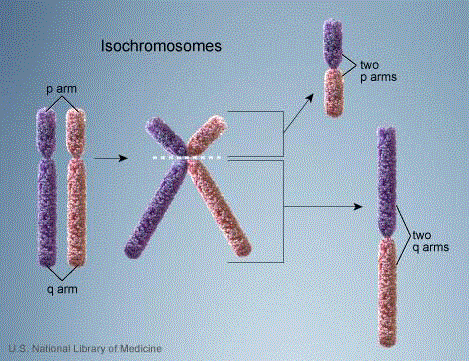
Figure 1
Formation of an isochromosome.
During either meiosis or mitosis, there is a transverse separation of
chromatids just above or below the centromere rather than the usual
longitudinal separation. The isochromosome consisting of the two short arms
(p) is usually lost. The remaining isochromosome consists of two long (q)
arms that are mirror images of each other.
Discussion
KS is characterized by heterogeneity in its clinical and genetic presentation [2,3]. Various phenotypic expressions have been described. The most common presenting phenotype is hypogonadism, gynecomastia, azoospermia or oligospermia, and increased levels of gonadotropins. The most common genotype is 47, XXY [2]. Genotypic variants of Klinefelter Syndrome are rare and usually consist of a multiplicity of normal X chromosomes in each cell, eg 48, XXXY or 49, XXXXY [5]. The occurrence of a supernumerary isochromosome of X is extremely rare. Only a few cases have been reported describing microsurgical testicular exploration and biopsy results in this rare genotype [6,7]. In addition, this patient’s normal phenotypic profile including normal virilization, normal serum testosterone concentrations and normal height is unusual. This subject’s laboratory testing indicating elevated FSH and LH suggest a lower likelihood of sperm retrieval by TESE which proved to be the case here. Karyotypes, however, are not always predictive of the likelihood of sperm retrieval in NOA. The sperm retrieval rate for patients with 46, XY karyotype, KS, and other chromosomal anomalies were 27.1%, 22.5%, and 15.4% respectively [6]. However, among the samples collected from the 13 patients with NOA due to chromosomal anomalies other than 47,XXY, only those from the two patients with the normal variant 46, XY,inv(9)(p12;q13) contained spermatozoa [6].
Conclusion
This case report adds to our current knowledge on the possibility of sperm retrieval by mTESE for NOA due to a supernumerary X isochromosome [8]. This case suggests that despite a normal phenotype and rare genotype, attempts at testicular sperm retrieval may be futile and reproductive planning may best be achieved using alternate approaches.
Authors' Contributions
Mia Letterie wrote the article. Janet L Kennedy, M.D. contributed suggestions for revision, the figure, and confirmed the references. R Dale McClure performed the microsurgical testicular sperm aspiration.
Acknowledgement
Gerard S Letterie, MD provided suggestions and encouragement to the first two authors. Thank you for your support.
References
- Plaseska-Karanfilska D, Noveski, P, Plaseski T, Maleva I, Madjunkova S, Moneva Z. Genetic Causes of Male Infertility. Balkan J Med Genet. 2012;15(Suppl):31-4.
- Smyth CM, Bremner WJ. Klinefelter syndrome. Arch Intern Med. 1998;158(12):1309-14.
- Bonomi M, Rochira V, Pasquali D, Balercia G, Jannini E, Ferlin A. Klinefelter syndrome (KS): Genetics, clinical phenotype and hypogonadism. J Endocrinol Invest. 2017; 40(2):123-34.
- Fedder J. Prevalence of small testicular hyperechogenic foci in subgroups of 382 non vasectomized, azoospermic men: a retrospective cohort study. Andrology. 2017;5(2):248-55.
- Tartaglia N, Ayari N, Howell S, D’Epagnier C, Zeitler P. 48, XXYY, 48, XXXY and 49, XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011; 100 (6):851-60.
- Takeda T, Iwatsuki S, Hamakawa T, Mizuno K, Kamiya H, Umemoto Y, et al. Chromosomal anomalies and sperm retrieval outcomes of patients with non obstructive azoospermia: A case series. Andrology. 2017;5(3):473-6.
- Demirhan O, Pazarbasi A, Tanriverdi N, Aridogan A, Karahan D. The clinical effects of isochromosome Xq in Klinefelter syndrome: Report of a case and review of literature. Genet Couns. 2009; 20(3):235-42.
- Arps S, Koske Westphal T, Meinecke P, Meschede D, Nieschlag E, Harprecht W, etal. Isochromosome Xq in Klinefelter syndrome: report of 7 new cases. Am J Med Genet. 1996;64(4): 580-2.