Case Report
Fryns Syndrome: Long Term Infertilty May Cause Teratogenicity/Mutation/Syndrome
Narayan JP1*, Karnawat BS1, Jain S1 and Narayan S2
1Department of Pediatrics, JLN Medical College, India
2Department of Obstetrics and Gynecology, LHMC, India
*Corresponding author: Jaiprakash Narayan Department of Pediatrics, Jawaharlal Nehru Medical College, JLN Medical College Circle, Near Kala Bag, Ajmer, Rajasthan 305001, India
Published: 12 Jun, 2017
Cite this article as: ValentinoNarayan JP, Karnawat BS, Jain
S, Narayan S. Fryns Syndrome:
Long Term Infertilty May Cause
Teratogenicity/Mutation/Syndrome. Ann
Clin Case Rep. 2017; 2: 1374.
Abstract
Fryns syndrome is syndrome in which Congenital Diaphragmatic Hernia (CDH) is a characteristic feature. Fryns syndrome comprises CDH and pulmonary hypoplasia, brachytelephalangy with nail hypoplasia, craniofacial dysmorphism, and organ malformations. We report a case of Fryns syndrome with all characteristics and after a long infertility without any treatment in elderly parents.
Keywords: Fryns syndrome; Elderly Parents; Infertility; Syndrome
Introduction
Fryns syndrome is the commonest autosomal recessive syndrome in which Congenital
Diaphragmatic Hernia (CDH) is a characteristic feature. FS comprises CDH and pulmonary
hypoplasia, brachytelephalangy with nail hypoplasia, craniofacial dysmorphism, orofacial clefting,
and organ malformations including cerebellar and neuronal heterotopias, ventricular septal defects,
renal cysts, and bicornuate uteri [1-3].
Diagnostic guidelines have been established but there are no formal diagnostic criteria for
FS or currently known biochemical or molecular markers for FS, and the aetiology has not been
determined. Several different chromosome aberrations have been associated with a phenotype
similar to FS [4,5]. For example, Pallister-Killian syndrome or tetrasomy 12p has frequently been the cause of a presentation resembling FS with CDH, pulmonary hypoplasia, coarse facial features,
aortic stenosis and cardiac septal defects, anal abnormalities, and hypoplasia of the external genitalia
[6]. CDH is associated with an underlying cytogenetic abnormality in 10%–33% of cases [4]. We report this case because our case is spontaneous conception product after a long infertility and elderly parents. It means long infertility and old age of parents may cause some genetic changes and
leads to syndromic clinical features in newborn.
Case Presentation
A newborn admitted in neonatal nursery at Govt. Mahila chikitsalaya Ajmer. He was delivered
as full term LSCS due to polyhydramnios and foetal distress. He had c/o difficulty in breathing,
sluggishness just after birth with apgar 6/10 at 1 min, 7/10 at 5 min. Age of mother at the time
of delivery was 45 years & fathers age 48 years. Age of mother at marriage was 20 years. She had
undergone D&C procedure at age of 22 years with 7 year treatment of infertility but not conceived.
At present document not available. She conceived at age of 45 years spontaneously without any
infertility treatment. Mother had taken tab iron folic acid, calcium during pregnancy. No history of
other medication. Second trimester USG shows polyhydromnios with multicystic kidney disease in
fetus Last week USG shows polyhydromnios with multicystic kidney disease in fetus.
Gestational age assessment by new ballard score is 38+-2 weeks. On examination weight, length
and head circumference were 2.8 kg, 43 cm, 32 cm. Child has scaphocephaly, large forehead, low
set ear, broad nasal bridge, hypertelorism, microphthalmia, narrow palpebral fissure, distorted
eyelashes, few hair at eyebrow, big nose, long philtrum, retognathia, high arched palate, short neck,
loose fold of skin at bask of neck, widely spaced nipple, abnormally large fingers and toes with
hypoplasia of nails, hypospadiasis, bilateral club foot and clinodactyly of 2nd toe bilaterally (Figure 1). Child has recurrent tearing with vomiting initially 3 days. Detail ophthalmic examination was
normal.
X-ray shows high left dome of diaphragm (Figure 2). Other findings in whole body x-ray were
normal. USG cranium was normal (Figure 3). USG abdomen and thorax shows eventration of diaphragm on left side with multicystic dysplastic kidney right side
(Figure 4). ECG was normal complete blood count within normal
limits. Other blood investigation were normal including CRP, urea,
creatinine, SGPT, sugar, blood culture.
Child was kept on O2 by hood for initial 36 hours. IV cefotaxime
was given for 7 days. Catori and spoon feed started at age of 48 hour.
Initially child has vomiting problem after each feed. Domperidone
drops and head end elevation done. Now child take catori and
spoon feed as well as mother feed well with no vomiting. Sample
for Chromosomal and genetic study was normal. Child discharged
successfully after 10 days of hospital stay. After 7 days on follow up
gained appropriate weight and well. 2d- echocardiography shows
complex congenital heart disease (ASD and multiple VSD). Later
on child admitted with breathing difficulty at age of 3 month with
diagnosis of pneumonia and successfully discharged. At age of 6
month child again of severe respiratory distress required ventilator
and dopamine, dobutamine support with diagnosis of severe
pneumonia, respiratory failure with shock and died.
Figure 1
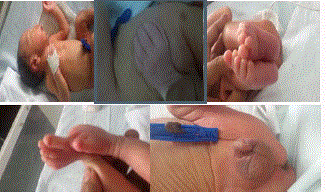
Figure 1
Physical examination of child.
Figure 2
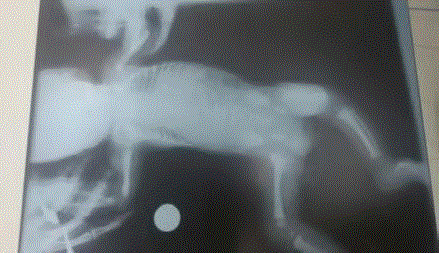
Figure 2
X-ray shows high left dome of diaphragm.
Figure 3
Figure 3
USG of cranium.
Figure 4

Figure 4
USG abdomen and thorax.
Discussion
Fryns syndrome is an autosomal recessive hereditary disease,
characterized by diaphragmatic defects (diaphragmatic hernia,
eventration, hypoplasia or agenesis); characteristic facial appearance
(coarse facies, ocular hypertelorism, broad and flat nasal bridge,
thick nasal tip, long philtrum, low-set and poorly formed ears, tented
upper lip, macrostomia, micrognathia); small thorax with widely
spaced hypoplastic nipples, distal digital hypoplasia (nails, terminal
phalanges); pulmonary hypoplasia; and associated anomalies
(polyhydramnios, cloudy corneas and/or microphthalmia, orofacial
clefting, renal dysplasia/renal cortical cysts, and/or malformations
involving the brain, cardiovascular system, gastrointestinal system, genitalia). Survival beyond the neonatal period has been rare. Data on postnatal growth and psychomotor development are limited;
however, severe developmental delay and intellectual disability are
common.
Fryns syndrome is the one of the most common syndromes
associated with Congenital Diaphragmatic Defect (CDH), reported
in up to 10% of patients with CDH. Although no eye abnormality was
seen in our patient, other findings were similar to the other typical
diagnostic findings. Fryns et al. [1] first described this syndrome
in 1979 with the major diagnostic criteria include abnormal facies
(coarse face, abnormal ear shape, cleft lip, cleft palate, large mouth,
microretrognathia, and broad nasal bridge), small thorax with
widely spaced hypoplastic nipples, distal limb and nail hypoplasia,
and diaphragmatic hernia with pulmonary hypoplasia [1,3].
Diaphragmatic hernia is a leading diagnostic feature in Fryns
syndrome, recorded in more than 80% of cases [4]. The rate of
mortality in the lethal phenotype has changed, considering the past,
and a 15% chance of survival is reported today [7]. Classically, Fryns
syndrome is characterized by distal limb hypoplasia. The spectrum of
distal limb hypoplasia includes short and broad hands, short digits,
short or absent terminal phalanges, hypoplastic or absent nails and clinodactyly [8]. Our patient showed short digits and nail hypoplasia.
Our case does not have diaphragmatic hernia, small thorax
pulmonary hypoplasia; and associated anomalies (cloudy corneas
and/or microphthalmia, and/or malformations involving the brain,
cardiovascular system, gastrointestinal system). Other characteristic
findings are similar to cases reported by other authors.
References
- Fryns JP, Moerman F, Goddeeris P, Bossuyt C, Van den Berghe H. A new lethal syndrome with cloudy corneae, diaphragmatic defects and distal limb deformities. Hum Genet. 1979; 50: 65–70.
- Pinar H, Carpenter MW, Abuelo D, Singer DB. Fryns syndrome: a new definition. Pediatr Pathol. 1994; 14: 467–478.
- Slavotinek AM. Fryns syndrome – a review of the phenotype and diagnostic guidelines. Am J Med Genet. 2004; 124A: 427–433.
- Enns GM, Cox VA, Goldstein RB, Gibbs DL, Harrison MR, Golabi M. Congenital diaphragmatic defects and associated syndromes, malformations, and chromosome anomalies: a retrospective study of 60 patients and literature review. Am J Med Genet. 1998; 79: 215–225.
- Lurie IW. Where to look for the genes related to diaphragmatic hernia?. Genet Couns. 2003; 14: 75–93.
- Veldman A, Schloesser R, Allendorf A, Fischer D, Heller K, Schaeff B, et al. Bilateral congenital diaphragmatic hernia: differentiation between Pallister- Killian and Fryns syndrome. Am J Med Genet. 2002; 111: 86–87.
- Neville HL, Jaksic T, Wilson JM, Lally PA, Hardin WD Jr, Hirschl RB, et al. Congenital Diaphragmatic Hernia Study Group. Fryns syndrome in children with congenital diaphragmatic hernia. J Pediatr surg. 2002; 37: 1685-1687.
- Bamforth JS, Leonard CO, Chodirker BN, Chitayat D, Gritter HL, Evans JA, et al. Congenital diaphragmatic hernia, coarse facies and acral hypoplasia: Fryns syndrome. Am J Med Genet. 1989; 32: 93-99.