Case Report
Aplasia Cutis Congenita of Scalp and Back: A Rare Entity
Virender Sekhon*
Department of Urology & Renal Transplantation, S.G Corporate Mobility Pvt Ltd, India
*Corresponding author: Virender Sekhon, Department of Urology & Renal Transplantation, S.G Corporate Mobility Pvt Ltd, Karnal, Haryana, India
Published: 05 Jun, 2017
Cite this article as: Sekhon V. Aplasia Cutis Congenita of Scalp and Back: A Rare Entity. Ann Clin Case Rep. 2017; 2: 1367.
Abstract
Aplasia cutis congenita of scalp is a rare, life-threatening birth defect. It may present as an isolated absent scalp skin, or with a combination of absent skull and absent skin in other body regions. We report a newborn with a very rare presentation of combination aplasia cutis congenita of scalp and lumbo-sacral region and discuss the differentials and management strategies.
Introduction
Aplasia cutis congenita is a rare, life-threatening birth defect having a circumscribed area of absent skin. 80-90% cases involve the scalp in a well demarcated and non-inflammatory fashion [1]. Of these, most are superficial involving only the epidermis and dermis but some are full-thickness
skin defects with absent epidermal appendages. 80% occur near the vertex and 20% are associated
with an underlying cranial bone defect [2]. Rarely, other regions of the body may be involved simultaneously.
We report a newborn with a very rare presentation of aplasia cutis congenita of scalp and
lumbo-sacral region.
Case Presentation
A female weighing 2.35 kgs was born by full-term vaginal delivery to a G3P2, 25 year old
mother. Antenatal period was uneventful and no record of any antenatal ultrasound was available.
The newborn had an 8 X 6 cms full thickness scalp defect with attenuated but intact dura, exposing
the underlying sagittal sinus. The head circumference was 35 cms. Another 3 X 1.5 cms area of
partial agenesis of the skin was present over the lumbo-sacral region (Figure 1A). Other systemic examination was essentially normal.
After prognostication and evaluation of surgical vs. non-surgical management options, parents
chose conservative treatment with moist gauze dressings and denied any further investigations,
including imaging.
Figure 1
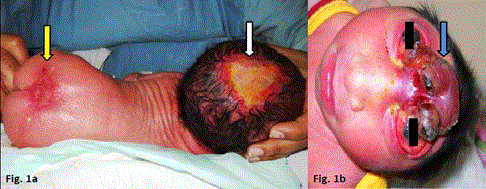
Figure 1
(A). Aplasia cutis congenital of scalp (white arrow) and lumbosacral
region (red arrow). (B). Anencephaly with absent scalp, skull and
forebrain (blue arrow) (Different patient).
Discussion
The pathophysiology of in utero skin disruption resulting in aplasia cutis congenita is yet
unexplained. The hypotheses include intrauterine vascular accidents, viral pathogens, pressure and
amniotic adhesions. Aplsia cutis can occur as a benign isoloated defect or as part of a syndrome,
sequence or association (eg. Goltz Gorlin syndrome, Tetrasomy 12p, Trisomy 13). Consanguinity,
maternal drug intake and genetic abnormalities (TGF-3 β-II receptor gene, BMS 1, EOGT missense
mutation) may explain the concurrent occurrence in scalp with other regions of the body [3]. Frieden’s classification for aplasia cutis congenita is based on the number and location of the lesions
and the presence or absence of associated malformations and consists of 9 groups [4].Mortality in cases of aplasia cutis congenital has been reported in 12 – 55% and it usually occurs during the first
week of life [5]. Of these, nearly 20% occur from bleeding. The exposed dura quickly becomes dry and dessicated, developing cracks which may spread into major dural veins causing exsanguinating
bleed [6]. Dural defects may also cause herniation of brain with mechanical injury. Local infection, meningitis, sepsis, CSF leak and thrombosis of superior sagittal sinus are the other challenges for
survival [3].
The treatment of this rare condition is controversial. Non-surgical management is preferred by
many for small defects with intact calvarium. This entails keeping the dura continuously moist with
either guaze dressing and saline drips or antiseptic ointments. The epithelialization is very gradual
and may take several months, during which the patient is at continued risk for complications [3].
Surgical management includes full-thickness rotational flaps covering the scalp defect in the first 24 hrs of life. Such aggressive intervention has been shown to
minimize hospital stay by reducing the time to secure wound closure.
It is therefore cost-effective with a low rate of complications and
appealing functional and aesthetic outcomes. Underlying dural
defect, when present, requires simultaneous closure with periosteal
flaps. However, calvarial defects do not always warrant a bone graft
as early scalp closure prompts ossification. Persistent bone defects
beyond 3-4 years age may need cranial reconstruction. Composite
closure of defects using bone and skin grafts in a single-stage has also
been described, but without much success.
Hydrocephalus is present in only a small percentage of patients
with aplasia cutis congenita. But intracranial hypertension may arise
from tight closure of dural or scalp defects or even a tight bandage
post-operatively. Growing brain of the child may exert undue force
on the undersurface of the flaps, causing delayed wound breakdowns
[3].
Aplasia cutis of the scalp can rarely be associated with lesions on
other parts of the body like face, trunk or limbs. To the best of our
knowledge, the association of a scalp with lumbo-sacral lesion has
not been previously described. These skin defects present in other
parts of the body are known to heal by spontaneous contraction
and epithelialization. However, a small percentage may have large
defects requiring primary closure with regional flaps or using tissue
expanding techniques [3]. A multi-disciplinary approach involving
neurosurgeons, pediatric surgeons and plastic surgeons is therefore
important in the care of these children.
Aplasia cutis congenita of the scalp must be distinguished from
anencephaly which is the congenital absence of scalp, skull and
forebrain (Figure 1B). A stage in the evolution of anencephaly is exencephaly or acrania characterized by complete or partial absence of skull bones, with complete but abnormal development of brain
tissue. Anencephaly results if the protruding brain of exencephaly
deteriorates. However, if it persists and is covered by skin or
epithelium, encephaloceles are formed. Another confusing term in
the same spectrum is mero-anencephaly (meroacrania) referring to
only a partial absence of brain and calvarium. Relatively small skull
and scalp defects may therefore be present in either least severe forms
of mero-anencephaly or most extreme forms of microcephaly with
encephalocele [7].
Conclusion
The index case represents a very rare combination of aplasia cutis congenital of scalp and lumbo-sacral region. A good knowledge of the condition and treatment options is essential for optimal case management. Multi-disciplinary team approach is required for evaluation of surgical versus non-surgical treatment, with the aim of protecting the brain from trauma and achieving long term aesthetic outcomes.
References
- Demmel U. Clinical aspects of congenital skin defects I. Congenital defects on the head of the newborn. Eur J Pediatr. 1975;121:21-50.
- Stephen MJ, Smith DW, Ponzi JW, Alden ER. Origin of scalp vertex aplasia cutis. J Pediatr. 1982;101:850-853.
- Winston KR, Ketch LL. Aplasia cutis congenital of the scalp, composite type: The criticality and inseparability of neurosurgical and plastic surgical management. PediatrNeurosurg. 2016.
- Freiden IJ. Aplasia cutis congenita: a clinical review and proposal for classification. J Am Acaddermatol. 1986;14:646-660.
- Dutra LB, Peretra MD, Kreniski TM, Zanon N, Cavalhetro S, Ferretra LM. Aplasia cutis congenital management of a large skull defect with acrania. J Craniofasc Surg. 2009;20:1288-1292.
- Ross DA, Laurie SW, Coombs CJ, Mutimer KL. Aplasia cutis congenital: failed conservative treatment. PlastReconstr Surg. 1995;95:124-29.
- Shewmon DA. Anencephaly: Selected medical aspects. The Hastings Center Report. 1988.