Case Report
Polyarteritis Nodosa in Association with Lactate Dehydrogenase Deficiency: A Case Report
Vivian Aranez, Parteet Sandhu and Julian L Ambrus*
Department of Medicine, SUNY at Buffalo School of Medicine, USA
*Corresponding author: Julian L Ambrus, Division of Allergy, Immunology and Rheumatology, SUNY at Buffalo School of Medicine, Room C281 BGH, 100 High Street, Buffalo, NY 14203 USA
Published: 19 Dec, 2016
Cite this article as: Aranez V, Sandhu P, Ambrus JL.
Polyarteritis Nodosa in Association with
Lactate Dehydrogenase Deficiency: A
Case Report. Ann Clin Case Rep. 2016;
1: 1214.
Abstract
Introduction: We describe a patient with a long history of fatigue and exercise intolerance who
became know to the Rheumatology service because of acute onset muscle pain and weight loss.
Muscle biopsy revealed Polyarteritis Nodosa (PAN), a type of rare systemic vasculitis predominantly
targeting medium sized arteries. Biochemical studies of the muscle revealed lactate dehydrogenase
deficiency and intermediate activity of carnitine palmitoyl transferase. The patient’s recovery
required treatment of both of the vasculitis and the metabolic disorder. We discuss how these
disorders could be interrelated.
Case Presentation: 61 year Caucasian male with a several year history of progressively worsening
fatigue and exercise intolerance who developed a 2-3 months history of generalized muscle weakness
and pain with weight loss. Laboratory evaluation and muscle biopsies demonstrated medium
size vessel vasculitis consistent with polyarteritis nodosa. Biochemical muscle biopsies revealed
diminished lactate dehydrogenase deficiency and low carnitine palmitoyl transferase activity.
Treatment consisted of steroids and cyclophosphamide for the vasculitis and for the metabolic
myopathy a low diet complex carbohydrates and ubiquinone, creatine, carnitine, folic acid, alpha
lipoic acid and ribose. The patient showed significant clinical improvement.
Conclusion: In our patient with symptoms of fatigue and exercise intolerance new symptoms of
muscle pain and weight loss revealed the development of a systemic vasculitis. Management of this
patient required treatment of both the underlying metabolic myopathy and the newly developed
systemic vasculitis. It is possible that having a metabolic myopathy predisposes to the development
of systemic vasculitis.
Keywords: Vasculitis; Lactate dehydrogenase deficiency
Case Presentation
A 61-year-old male presented with generalized muscle weakness associated with diffuse
muscular aches and pains, dyspnea on exertion, fevers, chills, fatigue and 20 pounds weight loss
which has been going on for one month. He was initially seen in Emergency Department (ED) and
labs revealed elevated troponin with WBC count of 20,000 with 36% Eosinophils, CPK of 1253 and
Creatinine of 0.87. Liver function test show AST and ALT 67, 68 respectively. Thyroid studies were
within normal limits. CRP was elevated at 72. 5, however, his ESR was at 32. Urinalysis obtained
showed 2+ blood, 0-2 RBC, trace glucose and 3+ ketones. Hepatitis panel A, B and C was negative.
His EBV IgM and CMV serology, ANA, CCP, RF, RNP and anti-smith antibody were negative. He
underwent coronary angiography that did not show any evidence of coronary artery disease. He
diagnosed with rhabdomyolysis and simvastatin was discontinued at discharge.
The patient again presented to ED one week post discharge with similar pain and generalized
weakness with no improvement after discontinuation of simvastatin. This time he was admitted
to the hospital for further evaluation. During this admission his WBC count was 15.2 with 12%
eosinophils, CPK was 404 and his troponin peaked at 4.68. He denied having chest pain but
complained of constipation, urinary retention and abdominal pain. He described the abdominal pain
as moderate, intermittent, sharp, and located in the peri-umbilical region. Abdominal ultrasound
showed hepatic steatosis and CT chest/abdomen/pelvis without contrast showed diverticulosis.
Differential diagnoses at the facility included inclusion body myositis, eosinophilic myositis, viral
myositis and statin induced myositis. The patient was started on Prednisone 40 mg PO once daily
and his weakness, myalgia and urinary retention improved. Patient was referred to Rheumatology
and Immunology clinic for further evaluation.
Presented at immunology clinic, he reported improved in
strength and energy, however he was still not back to his baseline. He
denied cough, wheezing, and shortness of breath at rest, hemoptysis,
sputum production, sensory deficits and any recent travels. His
allergic rhinitis and asthma remained controlled. He continued to
complaint dyspnea on exertion, constipation, 20-pound weight loss,
occasional fevers and chills.
His labs showed WBC count of 14, 300 with 0.1% eosinophils,
Hgb of 11.2, MCV of 82.2 and platelets of 615,000. He had ferritin
level of 1256, iron level of 21, transferrin saturation of 9% and
vitamin B12 level of 403. He had CK of 13 and creatinine of 1.08.
His ESR level increased to 84 and CRP decreased to 54. The patient
had normal C3, C4, IgA, IgG, IgM levels but IgE of 596. High ASCA
IgA and IgG were 21.7 and 81, respectively. P-ANCA was positive
at >1:512, C-ANCA and atypical P-ANCA were negative. He had a
low 25-hydroxy Vitamin D level of 24 and normal Carnitine level. He
had normal TSH and free T4. His gliadin Ab IgG and IgG and tissue
transglutaminase Ab IgG and IgA were normal.
CTA torso did not reveal any abnormalities. Muscle biopsy
was performed and showed a necrotizing vasculitis involving
medium sized muscular arteries and type 2 fiber atrophy (Figure 1).
Biochemical analysis of the muscle revealed normal myoadenylate
deaminase activity, very low lactate dehydrogenase activity(7.6
mcmol/min/g tissue, normal range 57.3 to 469 mcmol/min/g tissue;
3% of normal) and intermediately low carnitine palmitoyl transferase
activity (50.0 mcmol/min/g tissue, normal range 51.2 to 104.4 mcmol/
min/g tissue; 64% of normal).
The patient’s clinical history and muscle biopsy was consistent
with Polyarteritis nodosa (PAN) so he was treated with daily
cyclophosphamide initially at 1mg/kg and then adjusted to keep to
total WBC between 3-4,000/mL for 1 year along with prednisone 60
mg/day orally for 4 weeks and then tapered off over the next 3 months
[1,2]. For his metabolic myopathy he was placed on a diet low in
complex carbohydrates and rich in simple sugars with ribose 4000mg
4 times per day for his lactate dehydrogenase deficiency [3] and a
compound of CoQ10, creatine, carnitine, folic acid and alpha lipoic
acid for his carnitine palmitoyl transferase deficiency and secondary
mitochondrial dysfunction [4-6]. He showed dramatic improvement
in all his symptoms including fatigue, exercise intolerance, muscle
pain, abdominal pain and weight loss with 8-12 weeks. He returned
to work full time within 3 months.
Figure 1
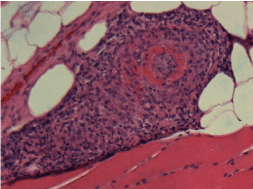
Figure 1
Muscle Biopsy.
Discussion
Polyarteritis Nodosa (PAN) is a type of rare systemic vasculitis
predominantly targeting medium sized arteries. In 2012, Chapel Hill
Consensus Conference (CHCC) define it as “Necrotizing arteritis of
medium or small arteries with glomerulonephritis or vasculitis in
arterioles, capillaries or venules and not associated with ANCA [7,8].
PAN encompass a spectrum of disorders, in which it maybe a systemic
disease or confined to single organ system. Clinically patients present
with fatigue, weight loss, myalgia and arthralgia. They typically have
mononeuritis multiplex, however they can also have symmetrical
polyneuropathy. It can cause cutaneous lesions like purpura, livedoid
lesions, subcutaneous nodules and necrotic ulcers. It does not cause
glomerulonephritis but it can cause renal tissue infarction and renal
micro aneurysms. Gastrointestinal tract can also be involved and is
usually associated with increased morbidity and mortality from the
disease [8].
In our case, the patient presented with weakness, myalgia and
weight loss, which is typical for PAN. Although the patient’s CTA
was negative, his muscle biopsy did show necrotizing obliteration
of medium size artery which was indicative of PAN (Figure 1). He
did have atypical findings such as elevated peripheral eosinophils,
however, he did not have features of asthma exacerbation or
uncontrolled allergic rhinitis putting eosinophilic granulomatosis
lower in the differential diagnosis [9]. Furthermore, the patient was
P-ANCA positive, while PAN is usually ANCA negative. However, in
the past there have been case reports published describing P-ANCA
positive PAN, some of these cases were confirmed with autopsies
[10,11].
In the past PAN was thought to be caused by circulating immune
complexes, but it is not associated with immune-complex mediated
glomerulonephritis or complement consumption [8]. In 1994 Cid et
al showed that T cells, particularly CD4+, play a major role in the
pathogenesis of PAN [12]. The etiology of PAN can be idiopathic
or can be triggered by viruses such as Hepatitis B virus, Hepatitis
C virus, Human Immunodeficiency virus, cytomegalovirus and
parvovirus B19 [13-16]. Adenosine deaminase 2 (ADA2) enzyme
deficiency has also been associated with PAN, perhaps because the
immunodeficiency made it harder for the patient to handle viral
infections [17,18]. In this case report, the patient had a complex
metabolic myopathy that also reduced his ability to handle viral
infections and therefore may have contributed to the development
of PAN [19-22].
Lactate Dehydrogenase is involved in glycolysis, especially in
anaerobic metabolism as it aids in conversion between pyruvate and
lactate. It consists of five isozymes and two subunits M and H. The H
subunit deficiency is clinically silent while the M subunit deficiency
leads to exercise intolerance and myopathy [23]. LDH deficiency
has not been linked previously with vasculitis, but we have observed
PAN in association with a mitochondrial myopathy and isomaltase
dysfunction [24]. Carnitine palmitoyl transferase deficiency is in
3-5% of the population and is also associated with fatigue and exercise
intolerance [25,26]. This patient had carnitine palmitoyl transferase
activity of 63% of normal so he could be heterozygote for carnitine
palmitoyl transferase deficiency. Whether or not this contributed
to his fatigue and exercise intolerance is unclear, but we chose to
treatment him for the mitochondrial dysfunction associated with
carnitine palmitoyl transferase deficiency at the same time that he
was treated for LDH deficiency. He did show dramatic improvement in his symptoms. In general, the medical community does need to be more aware of the existence of metabolic diseases and there potential
roles in various disease states [27,28].
References
- Cupps TR, AS Fauci. The Vasculitides. WB Saunders Company, Philadelphia. 1981; 21: 1-211.
- Forbess L, S Bannykh. Polyarteritis Nodosa. Rheumatic Disease Clinics of North America. 2015; 41: 33.
- Weinstein DA, JI Wolfsdorf. Glycogen storage diseases: A primer for clinicians. Endocrinol. 2002; 12: 531-538.
- Tarnopolsky MA. The mitochondrial cocktail: rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv Drug Deliv Rev. 2008; 60: 1561-1567.
- Angelini C, C Semplicini. Metabolic myopathies: the challenge of new treatments. Curr Opin Pharmacol. 2010; 10: 338-345.
- Abdullah M, S Vishwanath, A Elbalkhi, JL Ambrus. Mitochondrial myopathy presenting as fibromyalgia: a case report. J Med Case Rep. 2012; 6: 55.
- Jennette JC, RJ Falk, PA Bacon, N Basu, MC Cid, F Ferrario, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013; 65: 1-11.
- Hernandez-Rodriguez J, MA Alba, S Prieto-Gonzalez, MC Cid. Diagnosis and classification of polyarteritis nodosa. J Autoimmun. 2014; 48-49: 84- 89.
- Shimoi T, K Shojima, A Murota, Y Takizawa, J Maruyama, K Setoguchi. Clinical and pathological features of Churg Strauss syndrome among a Japanese population: a case series of 18 patients. Asian Pacific J Aller Immunol. 2012; 30: 61-70.
- Sugimoto T, K Kanasaki, T Koyama, Y Yokomaku, H Yasuda, A Kashiwagi, et al. A case of myeloperoxidase-antineutrophil cytoplasmic antibody positive-polyarteritis nodosa complicated by interstitial pneumonia and rapidly progressive renal failure. Clin Rheum. 2007; 26: 429-432.
- Tanaka M, K Matsuo, H Nakamura, S Ishikawa, K Matsuyama. [Two cases of classical polyarteritis nodosa associated with MPO-ANCA]. Nihon Jinzo Gakkai Shi. 2006; 48: 371-376.
- Cid MC, JM Grau, J Casademont, E Campo, B Collvinent, A Lopezsoto, et al. Immunohistochemical characterization of inflammatory cells and immunologic activation markers in muscle and nerve biopsy specimens from patients with systemic polyarteritis nodosa. Arthritis Rheum. 1994; 37: 1055-1061.
- Cohen P, L Guillevin. Vasculitis associated with viral infections. Presse Med. 2004; 33: 1371-1384.
- Cacoub P, B Terrier. Hepatitis B-Related Autoimmune Manifestations. Rheum Dis Clin North Am. 2009; 35: 125-137.
- Finkel TH, TJ Torok, PJ Ferguson, EL Durigon, SR Zaki, D Leung, et al. Chronic parvovirus B19 infection and systemic necrotising vasculitis: Opportunistic infection or aetiological agent? Lancet. 1994; 343: 1255- 1258.
- Meyer MF, B Hellmich, S Kotterba, H Schatz. Cytomegalovirus infection in systemic necrotizing vasculitis: causative agent or opportunistic infection. Rheumatol Int. 2000; 20: 35-38.
- Elkan PN, SB Pierce, R Segel, T Walsh, J Barash, S Padeh, et al. Mutant Adenosine Deaminase 2 in a Polyarteritis Nodosa Vasculopathy. New Eng J Med. 2014; 370: 921-931.
- Zhou Q, D Yang, AK Ombrello, AV Zavialov, C Toro, AV Zavialov, et al. Early- Onset Stroke and Vasculopathy Associated with Mutations in ADA2. New Eng J Med. 2014; 370: 911-920.
- van der Windt, GJ W, B Everts, CH Chang, JD Curtis, TC Freitas, et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8(+) T Cell Memory Development. Immunity. 2012; 36: 68-78.
- Walker MA, S Volpi, KB Sims, JE Walter, E Traggiai. Powering the Immune System: Mitochondria in Immune Function and Deficiency. J Immunol Res. 2014.
- Au WY, SC Chan. Association between glucose 6-phosphate dehydrogenase (G6PD) deficiency and fatal outcome of hepatitis E infection in middleaged men. Singapore Med J. 2012; 53: 148-149.
- Wendel U, H Schroten, S Burdach, V Wahn. Glycogen storage disease type Ib: infectious complications and measures for prevention. Eur J Pediatr. 1993; 152: S49-51.
- Kanno T, M Maekawa. Lactate dehydrogenase M-subunit deficiencies: clinical features, metabolic background, and genetic heterogeneities. Muscle Nerve Suppl. 1995; 3: S54-60.
- Vishwanath S, M Abdullah, A Elbalkhi, JL Ambrus. Metabolic myopathy presenting with polyarteritis nodosa: a case report. J Med Case Rep. 2011; 5: 262.
- Longo N, M Frigeni, M Pasquali. Carnitine transport and fatty acid oxidation. Biochimica Et Biophysica Acta-Molecular Cell Research. 2016; 1863: 2422-2435.
- Vladutiu GD. Genetic predisposition to statin myopathy. Curr Opin Rheumatol. 2008; 20: 648-655.
- Smith EC, A El-Gharbawy, DD Koeberl. Metabolic Myopathies: Clinical Features and Diagnostic Approach. Rheum Dis Clin North Am. 2011; 37: 201.
- Adler M, PB Shieh. Metabolic Myopathies. Seminars in Neurology. 2015; 35: 385-397.