Case Report
Dilated Cardiomyopathy in a 25-Year-Old Patient as a Result of Radiation and Chemotherapy after Stem Cell Transplantation
Raluca Diosteanu*, Gerhard Schuler and Hilka Gunold
Department of Internal Medicine and Cardiology, University Leipzig, Germany
*Corresponding author: Raluca Diosteanu, Department of Internal Medicine/ Cardiology, University Leipzig, Strümpellstraße 39, 04289 Leipzig, Germany
Published: 05 Dec, 2016
Cite this article as: Diosteanu R, Schuler G, Gunold H.
Dilated Cardiomyopathy in a 25-Year-
Old Patient as a Result of Radiation
and Chemotherapy after Stem Cell
Transplantation. Ann Clin Case Rep.
2016; 1: 1202.
Abstract
Dilated cardiomyopathy is a disorder characterized by chamber dilatation and cardiac dysfunction, which is not the result of an ischemic or valvular heart disease. The authors report the occurrence of dilated cardiomyopathy in an asymptomatic 25-year-old patient with a history of chemotherapy (Fludarabine and ATG- antithymocyte globuline) and radiation therapy prior to stem-cell transplantation for septic granulomatosis, which was diagnosed 10 years after transplantation. This proved to be refractory to the initiated medication and made the implantation of a CRT-D (cardiac resynchronization with defibrillation therapy) device necessary. The long-term outcome especially in young patients is unknown and the need for assist device or heart transplantation should be closely evaluated in case of further worsening of the cardiac function.
Keywords: Dilated cardiomyopathy; Chemotherapy; Fludarabine; ATG; Total body irradiation; Septic granulomatosis; Arterial hypertension; Cardiac resynchronization therapy
Introduction
Metastatic oral malignancy accounts for 1% of all oral cancers [1]. Primary sites of metastasis are lung, kidney, liver, breast, female genital organs and colo-rectum [2-3]. Metastasis from thyroid malignancies to this area is very rare. Papillary thyroid carcinoma favors lymphatic spread [4]. Hematogenous spread rarely occurs to the lung, bones and brain [5]. We report a rare case where metastasis from papillary thyroid carcinoma presented as an oropharyngeal mass, partially extending over wall of hypopharynx.
Case Presentation
A 25-year-old man was admitted to the department of internal medicine in November 2014 for
the management of a new diagnosed dilated cardiomyopathy with severely impaired ejection fraction.
The patient was asymptomatic at the time of diagnose. His history revealed arterial hypertension,
hyperlipidemia, overweight as well as septic granulomatosis (X-chromosomal hereditary). As a
result of the septic granulomatosis he suffered from many episodes of Aspergillus pneumonia (in
the Year 1996, 2000 and 2002), which led to resection of the left upper lobe (in the year 2000) and
of the inferior left lobe of the lung (in the year 2002). Afterwards, the patient received in 2004 stemcell
transplantation. Before transplantation a conditioning treatment involving chemotherapy with
Fludarabine, ATG (antithymocyte globulin) as well as total body irradiation was performed. The
last cardiologic follow-up was performed in January 2014. Back then a stress test showed no sign for
ischemia. Furthermore, the patient had a family history of cardiac disease (his uncle suffered from
sudden cardiac arrest and his grandfather had a pacemaker). The patient denied using alcohol and
drugs.
Actual physical examination revealed no signs of cardiac decompensation and the auscultation
provided physiological murmur sounds. The ECG (electrocardiography) at admission revealed a
sinus rhythm with narrow QRS complex. A transthoracic echocardiography showed an impaired
ejection fraction at 22% with global hypokinesia. Furthermore, a diastolic dysfunction II° was
reported (E>A, E/E’ 10). The diameter as well as the volume of the left ventricle (65 mm, respectively
220 ml) were considerably above the normal range (36-54 mm, respectively < 155 ml). The right
ventricular function was slightly reduced (TAPSE 16 mm).The performed coronary angiography
demonstrated a one-vessel coronary artery disease involving the left anterior descending artery,
in which a drug-eluting stent was implanted. Because of the complex medical history and unclear
etiology of the disease, we also performed a myocardial biopsy. The investigation revealed a chronic
myocardial damage due to an incipient dilated cardiomyopathy. Furthermore, there were signs of
a moderately active lymphocytic endocarditis without relevant inflammation in the heart muscle.
The patient was discharged with dual antiplatelet therapy (ASS and Clopidogrel) for 12 months
and optimized heart-failure therapy including an angiotensin-converting-enzyme inhibitor, a beta
blocker as well as an aldosterone inhibitor. To prevent sudden cardiac arrest the patient received a
Life Vest. Furthermore, in order to achieve a very close follow-up, the patient received telephone
monitoring over our special ambulance for heart failure. This way the heart-failure therapy was safely further optimized. Ivabradine, an inhibitor of the funny
channel, had to be discontinued because of adverse effects (burning
sensation of the tongue).The echocardiography at the next follow-up
in February 2015 revealed a minimal improvement of the ejection
fraction (30%) with no significant changes in size or volume of the
left ventricle (66 mm, respectively 233 ml). Furthermore, there was
also a reduction of the NT-pro-BNP (brain-type natriuretic peptide)
value from initially 4218 ng/l to 503 ng/l. Considering this slight
amelioration, a reevaluation of the CRT-D (cardiac resynchronization
with defibrillation therapy)was organized in2 months. The patient
had remained asymptomatic since admission. A genetic follow-up
was realized to search for inherited dilated cardiomyopathy even
though the results in April 2015 showed no evidence of mutations of
the LMNA (Lamin A/C), MYH7 (cardiac beta myosin heavy chain),
TNNT2 (cardiac troponin T) and MYBPC3 (cardiac myosin binding
protein C) genes.
On the next follow-up in April 2015, the patient presented with a
declined ejection fraction (EF 20%) as well as a higher NT-pro-BNP
value of 1044 ng/l. The patient denied the occurrence of any new symptoms. Considering the already optimized heart failure therapy
and the new wide total left bundle branch block (QRS 150 ms) with
pronounced asynchrony in the echocardiography, we decided to
move forward with the cardiac resynchronization by defibrillation
therapy (CRT-D), which was performed 3 weeks later. On the last
follow-up in October 2016 the echocardiography revealed no relevant
changes in size or volume of the left ventricle (65 mm, respectively
258 ml) despite the implantation of the CRT-D device with 100%
biventricular stimulation. The ejection fraction remained severely
impaired (EF 20%) and the NT-pro-BNP value was 893 ng/l.
In conclusion, after ruling out several other causes, we suspect
that the performed chemotherapy and total body irradiation are the
cause for the dilated cardiomyopathy in this very young patient.
Figure 1
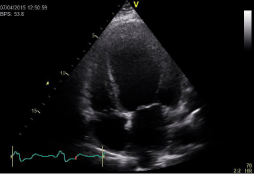
Figure 1
Dilated left ventricle with an ejection fraction of 22%.
Figure 2
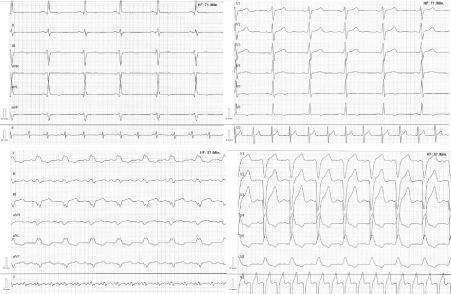
Figure 2
Electrocardiogram shows a sinus-rhythm with a narrow QRS complex (ECG from above) and a wide total left bundle branch block (150 ms, ECG from below).
Discussion
Dilated cardiomyopathy is a disorder characterized by chamber
dilatation, mostly left ventricle, and cardiac dysfunction, which is not
the result of an ischemic or valvular heart disease. Despite elaborate
investigations, in many patients there is no obvious cause for the
disorder, which is why most of them are assigned the diagnosis of
idiopathic dilated cardiomyopathy [1].
First of all, studies report several genetic mutations in a significant
percentage of cardiomyopathies, such as sarcomere DCM genes
(TNNT2, ß-MHC, MYBC3) which account for 35-40% of genetic
dilated cardiomyopathy, as well as Z band proteins and costamere
genes (MLP, CARP) or nuclear membrane defects in LMNA genes
[2,3]. In our case the genetic investigation presented no evidence of
mutations in the above mentioned genes making the familial dilated
cardiomyopathy unlikely. Secondly, a relevant differential diagnosis
could be arterial hypertension. Our patient received antihypertensive
Therapy since 2005, but the myocardial biopsy showed no specific
changes in this regard. Thirdly, another possible cause for the dilated
cardiomyopathy could be represented by the chemotherapy or radiation therapies, both of which our patient received 10 years earlier.
In long-time cancer-survivors with thoracic radiation, cardiovascular
disease is one of the leading causes of mortality, especially when
additional chemotherapy is performed. The side effects of the
radiation occurs mostly 10 to 20 years after therapy. Radiation therapy
leads to the generation of free radicals and DNA damage, leading to
endothelial dysfunction of the microvasculature, thrombosis and
small-vessel disease. Larger vessels, such as coronary arteries, can also
be affected, leading to stenosis [4]. Typical atherosclerosis gets along
only with intimal plaque formation, though, in radiation-induced
coronary artery disease it also comes to thinning of the media and
extensive adventitial fibrosis [5]. An approximate duration of 82
months to develop a radiation-induced coronary artery disease has
been described [6]. Myocardial damage induces progressive fibrosis,
diastolic dysfunction and finally restrictive cardiomyopathy [7].
Systolic dysfunction associated with radiation therapy alone, occurs
in less than 10% of the patients [8,9]. The relative risk of coronary
artery disease associated with radiation therapy increases with time
after exposure, at 10 years the risk of ischemia, sudden cardiac arrest
or heart failure is 2.9%, compared with 24.7% at 25 years [10]. This
complication is usually asymptomatic and difficult to diagnose, as
most of the patients do not experience any chest pain as a consequence
of impairment from radiation injury to sensory nerves in the chest
[11]. In our case, the patient denied angina.
Anthracycline-based chemotherapy and its cardiac toxicity are
well known. However, there are many chemotherapeutic agents in
which late cardiac complications have not been very well studied
and described. Our patient was treated with fludarabine and antithymocite-
globulin (ATG). In this regard our search through the
literature revealed no scientific evidence for cases of cardiac toxicity
related to these two drugs and future investigations in this area would
be required.
Lastly, autoimmune processes could play an important role in
developing cardiomyopathies. Autoantibodies directed at heart cell
receptors (muscarinic receptors, beta-adrenergic receptors as well
as myocyte proteins) have been described in the literature, although
the prevalence remains unclear [12]. The anti-ß1-adrenergic receptor
antibodies are suspected to promote cardiomyocyte apoptosis
leading to cardiac remodeling [13]. In our patient, the tumor
history including radiation and chemotherapy could have possibly
triggered a production of auto-antibodies; still there have been no
testing regarding this matter. Further studies in this area could raise
awareness of the importance of testing for auto-antibodies, which
could also improve the actual therapeutic strategies. In our case, the
diagnosis was made in an already advanced stage. The pump function
was refractory to medication, so that an additional therapy (cardiac
resynchronization with defibrillation therapy) was performed. The
long-term outcome is unknown, especially because of the young
age, and the patient should be monitored very closely in order to
initiate further investigations and therapy at an optimal time. At
this moment we don’t see the indication for an assist device or heart
transplantation.
In summary we emphasize the importance of further and
periodical evaluation, especially of young patients with a history of
chemotherapeutic and radiation therapy, because of its insufficiently
known side effects, especially its cardiac toxicity. Taking into
consideration the well-established side effects of the radiation therapy
we assume that an additional chemotherapy can aggravate and also
accelerate the process of cardiac damage. Clinical examination and
trans-thoracic echocardiography should be regularly performed,
in order to early diagnose cardiac dysfunctions and allow proper
treatment on time.
References
- Falk RH, Hershberge RE, editors. The Dilated, Restrictive and Infiltrative Cardiomyopathies. Philadelphia. Elsevier Saunders. 2015; 1551 (Braundwald E, Mann DL, Zipes DP, Libby P, Bonow RO, editors. Braunwald’s Heart Disease. A text book of cardiovascular medicine; vol. 2)
- McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013; 123: 19-26.
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012; 366: 619-628.
- Chen MH, Force T, editors. Cardiovascular Complications of Cancer Therapeutic Agents. Philadelphia. Elsevier Saunders. 2015. 1620-1621 (Braundwald E, Mann DL, Zipes DP, Libby P, Bonow RO, editors. Braunwald’s Heart Disease. A textbook of cardiovascular medicine; vol 2)
- Virmani R, Farb A, Carter AJ, Jones RM. Pathology of radiation-induced coronary artery disease in human and pig. Cardiovasc Radiat Med. 1999; 1: 98-101.
- Veinot JP, Edward WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol. 1996; 27: 766-773.
- Yusuf SW, Sami S, Daher IN. Radiation-induced heart disease: a clinical update. Cardiol Res Pract. 2011; 2011: 317659.
- Adams MJ, Lipsitz SR, Colan SD, Tarbell NJ, Treves ST, Lisa D, et al. Cardiovascular status in long-term survivors of Hodgkin’s disease treated with chest radiotherapy. J Clin Oncol. 2004; 22: 3139-3148.
- Heidenreich PA, Hancock SL, Lee BK, Mariscal CS, Schnittger I. Asymptomatic cardiac disease following mediastinal irratiation. J Am Coll Cardiol. 2003; 42: 743-749.
- Glanzmann C, Kaufmann P, Jenni R, Hess OM, Huguenin P. Cardiac risk after mediastinal irradiation for Hodgkin’s disease. Radiother Oncol. 1998; 46: 51-62.
- Chen MH, Colan SD, Diller L. Cardiovascular disease: Cause of morbidity and mortality in adult survivors of childhood cancers. Circ Res. 2011; 108: 619-628.
- Dandel M, Wallukat G, Potapov E, Hetzer R. Role of ß1-adrenoceptor autoantibodies in the pathogenesis of dilated cardiomyopathy. Immunobiology. 2012; 217: 511-520.
- Jane-wit D, Altuntas CZ, Johnson JM, Yong S, Wickley PJ, Clark P, et al. Beta 1-adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation. 2007; 116: 399-410.