Case Report
An Exceptional GPA: Granulomatosis with Polyangiitis Presenting as Hypertrophic Pachymeningitis
Mazzella AJ1*, Kelly M2, Falk R1, Jeanette JC3 and Szpak C4
1Department of Internal Medicine, University of North Carolina Hospitals, USA
2Department of Hospital Medicine, Wake Med Health and Hospitals, USA
3Department of Pathology, University of North Carolina Hospitals, USA
4Department of Pathology, Wake Med Health and Hospitals, USA
*Corresponding author: Anthony J. Mazzella, Department of Internal Medicine, University of North Carolina Hospitals, 125 MacNider Hall / Campus Box #7005/ Chapel Hill NC 27599-7005, USA
Published: 28 Jul, 2016
Cite this article as: Mazzella AJ, Kelly M, Falk R, Jeanette
JC, Szpak C. An Exceptional GPA:
Granulomatosis with Polyangiitis
Presenting as Hypertrophic
Pachymeningitis. Ann Clin Case Rep.
2016; 1: 1051.
Abstract
Background: Granulomatosis with polyangiitis (GPA) is a necrotizing vasculitis usually involving
small blood vessels that has been described to rarely effect the meninges, presenting with headaches
and in our case papilledema. Thickened dura mater may be seen on brain imaging and is an entity
known as hypertrophic pachymeningitis. We present a case of isolated dural recrudescence of
ANCA vasculitis.
Case Presentation: A 62-year-old man with known anti-GBM disease and history of ANCA
positivity who remains dialysis dependent after initial diagnosis and treatment eight months
prior presents to the emergency department from his ophthalmologist’s office due to headaches,
vision changes, and severe bilateral papilledema. Initial head imaging with computed tomography
revealed hypertrophic pachymeningitis and lumbar puncture revealed elevated opening pressure
of 38 cm H2O. Further workup was unremarkable, including persistently negative ANCA titers.
Ultimately dural biopsy led to diagnosis of GPA with isolated dural involvement, likely representing
recrudescence from his original diagnosis eight months prior. He was started on IV steroids,
underwent plasmapheresis, and started on rituximab with symptomatic improvement.
Conclusions: Hypertrophic pachymeningitis is a rare phenomenon that can be caused by GPA.
Prompt recognition and treatment of increased intracranial pressure is essential to avoid vision loss
and life-threatening complications.
Keywords: ANCA; Anti-GBM disease; Vasculitis; Hypertrophic pachymeningitis; Papilledema
Abbreviations: GPA: Granulomatosis with Polyangiitis; ANCA: Anti-Neutrophil Cytoplasmic Antibody; Anti-GBM: Anti-Glomerular Basement Membrane; ICD: Implantable Cardiac Defibrillator; MPOANCA: Myeloperoxidase Anti-Neutrophil Cytoplasmic Antibody; MRI: Magnetic Resonance Imaging; CT: Computed Tomography; AFB: Acid-Fast Bacilli
Background
Granulomatosis with polyangiitis (GPA) is necrotizing granulomatous vasculitis characterized by anti-neutrophil cytoplasmic antibody (ANCA) positivity which most often affects the nephrons and/or small airways [1]. Limited presentations can include other regions with vessels of similar caliber including the meninges leading to dural thickening. Hypertrophic pachymeningitis is a radiologic finding that refers in an inflammatory thickening of the dura mater which can contribute to increased intracranial pressure or cranial nerve palsies. We present a rare case of isolated dural ANCA vasculitis.
Case Presentation
A 62-year-old African American male with history of rapidly progressive glomerulo nephritis
due to anti-glomerular basement membrane (anti-GBM) disease who remains dialysis dependent
after failing to respond to steroids and plasmapheresis eight months ago presented to the emergency
department with two months of worsening vision and frontal headaches. The patient also has
chronic systolic heart failure with an implantable cardiac defibrillator (ICD). Further history was
remarkable for positive myeloperoxidase anti-neutrophil cytoplasmic antibody (MPO-ANCA) at the time of his initial anti-GBM diagnosis. Both ANCA and anti-GBM titers have since been undetectable and there have been no reported symptoms of recrudescent disease. He was not on maintenance immunosuppressive therapy as he remained dialysis dependent and was otherwise asymptomatic.
For the last two months or so the patient has been experiencing progressive vision changes leading to intermittent vision loss which he describes as a gradual dimming and seeing dark shadows in all visual fields. The patient saw his outpatient ophthalmologist who observed severe bilateral papilledema prompting referral to the emergency department.
Further review of systems revealed approximately 40-pound unintentional weight loss as well as increased fatigue over the last three-to-four months. He denied fever, skin rashes, joint pain, rhinorrhea, sinus pressure, shortness of breath, chest pain, or cough.
Current outpatient medications included amlodipine, carvedilol, valsartan, hydralazine, and calcium acetate. He has been fully compliant with his three times weekly dialysis sessions.
On initial examination, the patient’s blood pressure was 170/101 mmHg, pulse was 90 beats per minute, oral temperature 98.2°F, and his oxygen saturation was 97% while breathing ambient air. His height was 168cm, weight 82kg, and body-mass index 29 kg/m2. He appeared mildly uncomfortable but did not exhibit light sensitivity. His pupils were equal, round, and reactive to light bilaterally. Extra ocular movements were intact. He had bilateral papilledema on funduscopic examination. There were no cranial nerve deficits and his visual fields were intact. The remainder of the physical exam was normal. The white blood cell count, hemoglobin, platelet count, electrolytes, liver function, and tests of coagulation were unremarkable for a patient on hemodialysis.
As a consequence of headaches and visual disturbances, imaging of the brain was required. Magnetic resonance imaging (MRI) with venogram was preferred given papilledema however this possibility was excluded due to the patient’s ICD. Instead, computed tomography (CT) of the brain was obtained with CT venogram which revealed dural thickening concerning for hypertrophic pachymeningitis (Figure 1) with diminished caliber of the superior sagittal and straight sinuses. Lumbar puncture revealed elevated opening pressure of 38 cm H2O, 10 WBC/μL, 3401 RBC/μL, protein 137 mg/dL, and glucose 64 mg/dL. He experienced temporary symptomatic improvement on acetazolamide while further work-up was pending. Extensive CSF studies including gram stain with culture, cytology, acid fast bacilli (AFB) smear, rapid plasma reagin, cryptococcal antigen, and angiotensin converting enzyme were negative. Serum anti-nuclear antibody and ANCA titers were negative as were blood cultures, serum AFB, and HIV.
The first several days of hospitalization were spent obtaining these results and the patient’s headache was temporized with acetazolamide with diminishing efficacy. The hypertrophic pachymeningitis on brain imaging includes a wide differential diagnosis such as sarcoidosis, syphilis, lymphoma, subarachnoid hemorrhage, tuberculosis, and ANCA vasculitis. Given the wide spectrum of possible etiologies with unrevealing work-up, and potential detriment from treatment if the wrong diagnosis was pursued, a dural biopsy was performed. Our neurosurgical colleagues remarked that the patient’s dura was 3-4 times normal thickness and grossly granulomatous in appearance. Pathologic examination revealed necrotizing vasculitis involving the small vessels with granulomatous inflammation concerning for granulomatosis with polyangiitis (GPA) (Figure 2 and 3). The patient was started on IV steroids, plasmapheresis, and rituximab. He experienced symptomatic improvement of his headaches and vision changes shortly thereafter.
Figure 1
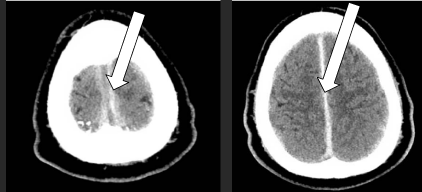
Figure 1
CT scan revealing hypertrophic pachymeningitis: rare condition involving inflammatory thickening of the dura mater (arrows).
Figure 2
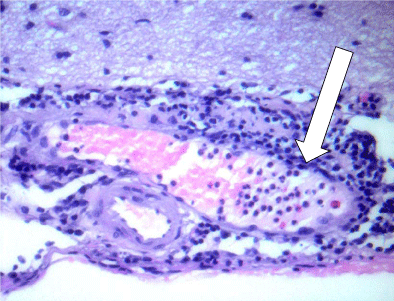
Figure 2
Segmental vasculitis with marginating neutrophils in a small vein (arrow) in the subarachnoid space with adjacent meningeal inflammation.
Figure 3
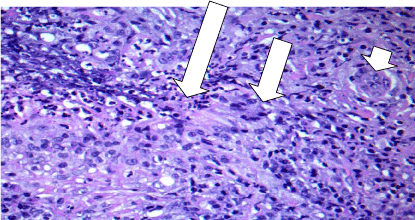
Figure 3
Necrotizing granulomatosis with central zone of necrosis and neutrophils (long arrow), adjacent macrophages (middle arrow), and including multinucleated giant cells (short arrow).
Discussion
Inflammation of the central nervous system is a rare but documented manifestation of GPA. In two case series from the 1990’s, meningitis was observed in two out of a combined total of 482 patients (0.4%) [2,3]. A review from 2006 found 46 additional cases, three of which appeared to have isolated dural involvement [4]. Demographics data from this review shows a male/female ratio of 1.13, with mean age at diagnosis of 48.2 ± 15 years. The mean time between symptom onset and diagnosis of meningitis varied widely and was about 35 ± 60 months with a range between 0-240 months. Thirty-seven of these 48 cases included detailed clinical information with most common symptoms being headache and cranial nerve palsies usually in the absence of meningeal signs. About two thirds of patients had positive serum ANCA at the time of diagnosis of meningeal involvement. Twenty-six patients underwent meningeal biopsy with the most common histopathologic finding being necrotizing granulomatosis without vasculitis (62%). Approximately 15% of patients had both necrotizing granulomatosis and small vessel vasculitis as with our case.
Hypertrophic pachymeningitis is a rare condition involving inflammatory thickening of the dura mater which may lead to signs of increased intracranial pressure or cranial nerve palsies. MRI is the diagnostic modality of choice to identify meningeal thickening [5]. CT scan has limited sensitivity but can still be of diagnostic utility as in this case. Two patterns of hypertrophic pachymeningitis have been described: diffuse and focal, which has led to postulation of disease mechanisms. The focal form has a predilection to the paranasal and periorbital regions which may be a result of direct extension of granulomatous inflammation from the nasal cavities reaching the meninges. The more diffuse form, as in our patient, may represent a primary presentation or recrudescence of the disease but is poorly understood [1,5].
Another interesting feature of this case is the original anti-GBM diagnosis eight months prior to this presentation. This case illustrates an example of anti-GBM disease with MPO positive ANCA which is a disease entity associated with worse renal prognosis than anti-GBM disease or ANCA vasculitis alone [6]. Clinicians should consider maintenance therapy in these patients even if ANCA titers turn negative, as negative titers do not prove dormancy of underlying vasculitis [1].
Conclusion
Hypertrophic pachymeningitis is a rare phenomenon that can be caused by GPA. Prompt recognition and treatment of increased intracranial pressure is essential to avoid vision loss and life-threatening complications. Typically, there are systemic manifestations of disease recrudescence, often times including more accessible tissue for biopsy. If that is not the case, a dural sample may be necessary to facilitate definitive and prompt diagnosis thus guiding therapy. Patients with meningeal involvement often demonstrate clinical improvement with steroids and immunosuppressive medications despite minimal changes radiographically [7,8].
This case reminds the clinician that negative ANCA titers do not disprove recrudescence of the disease, and that vasculitis can have localized presentations in very unusual places.
Authors' Contributions
AM was the main author of the manuscript. MK was a contributing author of the manuscript. RF was a contributing author of the manuscript and contributed his expert opinion in the field. CJ confirmed the histopathologic diagnosis and contributed his expert opinion in the field. CS performed the initial histopathologic examination of the dura mater and initially confirmed the diagnosis. All authors have reviewed the manuscript.
References
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. “2012 revised International Chapel Hill consensus conference nomenclature of vasculitides,” Arthritis & Rheumatism. 2013; 65: 1-11.
- Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992; 116: 488-498.
- Nishino H, Rubino FA, DeRemee RA, Swanson JW, Parisi JE. Neurological involvement in Wegener’s granulomatosis: An analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. 1993; 33: 4-9.
- G Di Comite, Bozzolo EP, Praderio L, Tresoldi M, Sabbadini MG. “Meningeal involvement in Wegener’s granulomatosis is associated with localized disease,” Clinical and Experimental Rheumatology. 2006; 24: S60-S64.
- Murphy JM, Gomez-Anson B, Gillard JH, Antoun NM, Cross J, Elliott JD, et al. “Wegener granulomatosis: MR imaging findings in brain and meninges,” Radiology. 1999; 213: 794-799.
- Levy JB. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int. 2004; 66:1535-1540.
- Sakellariou GT, Kefala N. Pachymeningitis in Granulomatosis with Polyangiitis: A Case Report and a Review of the Literature. Case Reports in Rheumatology. 2013; 840984.
- Fam AG, Elana L, Liesly L, Bayardo PO, Mayank G. “Cranial pachymeningitis: an unusual manifestation of Wegener’s granulomatosis,” Journal of Rheumatology. 2003; 30: 2070-2074.