Case Report
Kinsbourne Syndrome as the Unique Manifestation in Children with Neuroblastoma: Case Report
Sabbaga CC*, Antunes LA, Talini C, Neves de Carvalho CB, Bersani Amado FA and Aranha
Junior AA
Department of Pediatric Surgery, Hospital Pequeno Príncipe, Brazil
*Corresponding author: César Cavalli Sabbaga, Department of Pediatric Surgery, Hospital Pequeno Príncipe, Curitiba-PR, Brazil
Published: 07 Jul, 2016
Cite this article as: Sabbaga CC, Antunes LA, Talini C,
Neves de Carvalho CB, Bersani Amado
FA, Aranha Junior AA. Kinsbourne
Syndrome as the Unique Manifestation
in Children with Neuroblastoma: Case
Report. Ann Clin Case Rep. 2016; 1:
1042.
Abstract
The opsoclonus-myoclonus-ataxia syndrome (OMAS) also known as Kinsbourne syndrome can occur as a single neurological event in children with paraneoplastic syndrome in a neuroblastoma low degree. This study aims to report a Kinsbourne syndrome in a 3 year-old-child and a literature review focused on the diagnosis and therapy alternatives. This report emphasizes the need for neurologists and pediatricians to suspect the hypothesis of an indolent neuroblastoma in patients presenting OMAS with no neuroimaging study changes. The high association with neuroblastoma is reported and should not be excluded even if a first investigation is negative. The patients high percentage with neurocognitive sequelae and permanent deficits is a challenge in the search for new and more aggressive treatment options.
Keywords: Kinsbourne syndrome; Neuroblastoma; Opsoclonus-myoclonus-ataxia syndrome
Introduction
The opsoclonus-myoclonus-ataxia syndrome (OMAS) also known as Kinsbourne syndrome
can occur as a single neurological event in children with paraneoplastic syndrome in a low degree
neuroblastoma [1,2]. The syndrome occurs most frequently in young children - 3 to 6 years old - and
accounts for 2 to 3% of children with neuroblastoma, with an estimated incidence of 1:5 million. It is
presented with rapid, irregular, involuntary, horizontal and vertical eye movements and there is no
intersaccadic interval (opsoclonus), myoclonus, irritability and cerebellar ataxia [3].
Fifty percent of patients have an indolent neuroblastoma [4]. This syndrome manifests itself
in small tumors (stage I or II) with favorable histology and without N-myc gene amplification.
These tumors rarely recur after resection and the survival is higher than children who do not have
neurologic manifestations. Other causes would be idiopathic, associated with viral infections,
metabolic and autoimmune diseases [2,4].
It is accepted that the neurological paraneoplastic syndrome occurs when there are similar
antigens between tumor cells and neurons [1]. Despite treatment with corticosteroids, corticotropin
(ACTH) and intravenous immunoglobulin, many children with ataxia maintain a residual deficit
even after the tumor resection and in some cases there may be severe cognitive deficits as OMAS
sequel [2].
This report aims to describe the case of a 3-year-old child who presented OMAS as the unique
manifestation of an indolent neuroblastoma, focusing in the diagnostic and therapeutic update.
Case Presentation
A 3 years old, female, was presented to the pediatric neurologist with these symptoms for 3
months: dysarthria when irritated, opsoclonus, gait ataxia, trunk and head pole, dysmetria and
diadochokinesia and no previous diagnosis in her origin city. The patient was born at term,
weighted 3450 grams. The family history without change. A neurodevelopment was held on her
head when she was 4 months old, sat with 6 months, walked with 1 year and 20 days, she spoke her
first words with 9 months. A MRI brain and cerebrospinal fluid was performed with no alteration.
An electroencephalogram showed a background activity revealing slow-wave outbreaks in the
widespread projection with unspecific aetiological nature. The laboratory tests to Vitamin D, B12,
B6, E, LDH and ferritin were normal. The vanillylmandelic acid in urine was 2.5 mg / 24 hours
(normal value from 2.0 to 14 mg / 24 hours).
The abdominal ultrasound was normal but a CT scan was also performed and it revealed expansive adrenal disease on the right measuring 29 x 14 x 21 mm with hyperdense image suggesting calcification (Figure 1 and 2). The chest CT was without lesions.
With the presumptive neuroblastoma diagnosis, the patient underwent a right adrenalectomy. The procedure was uneventful. The histopathologic analysis was consistent with adrenal neuroblastoma with low mitotic index - less than 4% - (Stage I). The myelogram and bone marrow biopsy were without alteration. The N-myc gene was negative. In the postoperative period an intravenous immunoglobulin use was started with a good response, discreet permanent ataxia and periods of aggression.
After ten months of clinical follow up with no medication use, a new febrile episode with opsoclonus and ataxia without tumor recurrence was presented (Figure 3). Immunoglobulin was restarted at a dose of 0.4 mg / kg / day for 5 days and dexamethasone 0.33 mg / kg / day. She was discharged in use of oral steroid (prednisone 1 mg / kg / day). She remains in follow up with the pediatric oncology and has presented no symptoms so far, without new acute crisis episodes for one year.
Figure 1
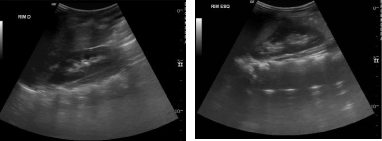
Figure 1
Abdominal ultrasound with alteration absense.
Figure 2
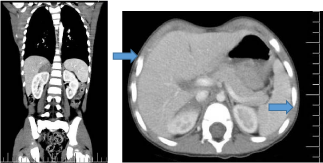
Figure 2
The CT revealed an expansive adrenal disease on the right measuring 29 x 14 x 21mm with hyperdense image suggesting calcification.
Figure 3
Figure 3
Imaging tests show no recurrence of neuroblastoma.
Discussion
In order to diagnose OMAS the presence of at least three out of four criteria is needed: opsoclonus, ataxia and / or myoclonus, behavioral disorders and neuroblastoma [1]. Neuroblastomas associated with OMAS are generally small, well differentiated, localized and less than 10% are metastatic. It is usually associated with high survival rate when compared to similar tumors in children without symptoms associated with OMAS [4].
Aguilera et al. [3], reported that Kinsbourne syndrome may precede in months to years the appearance of tumors of the neural crest, so there must be suspicion in order to perform radiological investigation. First line exams are chest X-ray and abdominal ultrasound. If normal, the investigation must proceed with abdominal CT scan or MRI as in the present case [3]. OMAS syndrome may occur several months before any biochemical evidence or tumor image [5].
Nuclear medicine studies, preferably 123I-meta-iodobenzylguanidine (MIBG) scintigraphy examination, are essential during the initial phase to establish diagnosis and to search for distant metastasis, particularly bone metastasis. Alternatively, 131I-MIBG or technetium 99m (TC) methyldiphosphonate scintigraphy may be used for the same purpose. Nuclear medicine studies are also used to follow response to therapy. Follow-up imaging is required to assess for residual tumor after surgery, response to chemotherapy, to decide possible operability, to assess tumor recurrence, and to search for distant metastasis [6].
Olbere et al. [4] reported the delay to diagnose neuroblastoma with a median of three months, as in this case, due to the unique neurological manifestation prior to chest or abdominal radiological symptoms and investigation.
The presence of autoantibodies to neurons and Purkinje cells can be detected. Some of these autoantibodies can be recognized and establish specifically the neuronal surface antigens and intracellular neuroblastoma cell lines, inhibiting proliferation and inducing apoptosis. Thus, it is speculated that the neuroblastoma may spontaneously regress before the detection of opsomioclonus [4].
Due to the suspicion of the immune pathogenesis, the treatment is based in corticosteroids and in immunomodulatory therapy such as cyclophosphamide, azathioprine and plasmapheresis. Some recent studies cite rituximab in patients refractory to previous treatments [1,7]. Tumor resection can improve the acute symptoms for a short period but most require immunosuppressive treatment. Reccurency is frequent due to discontinuation of treatment or environmental triggers, as shown in the case [3]. Therapies with corticosteroids or adrenocorticotropic hormone (ACTH) are beneficial to control the neurological symptoms in recurrent forms of the disease and are usually the first treatment option. In some cases maintenance therapy is needed for several months as happened to our patient that after the second exacerbation crisis was discharged using oral corticosteroids therapy [4]. Table 1 shows options in therapeutic management OMAS [8].
In 70-80% of cases patients remain with neuropsychological sequelae, regardless of whether associated or not with neuroblastomas [1]. The motor and cognitive consequences are the main difficulty in these patients management [1,3,5]. Only a minority of children have a monophasic course and better prognosis [4]. Manifestation as behavioral disorders, cognitive, language and learning, sleep disorders and psychiatric problems of order are commom.
Early treatment with imunosupresormight lead to better prognosis decreasing the percentage of neuropsychological sequelae. With or without treatment, opsoclonus is usually reverted but minimum eye movement abnormalities, jerky movements during the search may persist even years after treatment. After treatment motor function generally improves and patients are able to walk individually. This improvement in motor function is slow, however, cognitive disorders in the area are usually permanent. If untreated the motor deficit remains unchanged. Some of them present difficulties in learning and language. Neurological symptoms may fluctuate during treatment with frequent relapses decreases when immunosuppressive treatment or concurrent viral diseases [1].
Table 1
Table 1
Study variables measured during the stationary intensive rehabilitation.
Conclusion
This report emphasizes the need for neurologists and pediatricians to suspect the hypothesis of an indolent neuroblastoma in patients presenting OMAS with no changes in brain CT. The high association with neuroblastoma is reported and should not be excluded even if a first investigation is negative. The high percentage of patients with neurocognitive sequelae and permanent deficits is a challenge in the search for new and more aggressive treatment options.
References
- Bravo J, Lopez-Almaraz R, Mateos M, Diaz L, Hernandez-Exposito S. [Neuropsychological profile in opsoclonus-myoclonus-ataxia syndrome presenting as neuroblastic tumours]. Rev Neurol. 2016; 62: 249-257.
- Mitchell WG, Gonzalez YD, Brumm VL, Aller SK, Burger E, Turkel SB, et al. Opsoclonus-Ataxia caused by childhood neuroblastoma: Developmental and neurologic sequelae. Pediatrics. 2002; 109; 86-98.
- Aguilera Albesa S, Botella MP, Salado C, Bosque A, Ocio I, Montiano JI. [Paraneoplastic opsoclonus myoclonus ataxia syndrome]. An Sist Sanit Navar. 2009; 32: 91-95.
- Andrade OVB, Troster EJ, Cypel S. Kinsbourne syndrome manifesting with signs of post-viral encephalitis. Rev Paul Pediatr. 2011; 29: 300-304.
- Dávila-Gutierrez G, Palacios-Acosta JM, Guzmán-Martinez A, Rodríguez-Abarca F, Shalkow-Klincovstein J, Carrasco-Daza D. Síndrome de opsoclonos mioclonos causado por neuroblastoma. Informe de um caso. Acta Pediatr Mex. 2010; 3: 36-41.
- Balassy C, Navarro OM, Daneman A. Adrenal masses in children. Radiol Clin North Am. 2011; 49: 711-727.
- Tate ED, Pranzatelli MR, Verhulst SJ, Markwell SJ, Franz DN, Graf WD, et al. Active Comparator-Controlled, Rater-Blinded Study of corticotropin-Based Immunotherapies for Opsoclonus-Myoclonus Syndrome. J Child Neurol. 2012; 27: 875-884.
- Arroyo HA, Tringler N. [Opsoclonus-myoclonus syndrome]. Medicina (B Aires). 2009; 69: 64-70.