Case Report
Small Pheochromocytoma in an Elderly Woman with Hypertension
Minamoto M1, Fukuoka T1*, Umakoshi H1 and Murakami K2
1Department of Internal Medicine, Matsuyama Red Cross Hospital, Japan
2Departmetnt of Health care and Preventive Medicine, Matsuyama Red Cross Hospital, Japan
*Corresponding author: Tomikazu Fukuoka, Department of Internal Medicine Ichi-banchi, Matsuyama Red Cross Hospital, Bunkyou-tyou, Matsuyama-shi. Ehime, 790-8524, Japan
Published: 02 Jul, 2016
Cite this article as: Minamoto M, Fukuoka T, Umakoshi H,
Murakami K. Small Pheochromocytoma
in an Elderly Woman with Hypertension.
Ann Clin Case Rep. 2016; 1: 1038.
Abstract
A 77-year-old woman who had controlled hypertension was referred to our hospital due to recent paroxysmal elevation in blood pressure. Computed tomography showed a 17-mm nodule in right adrenal gland, whereas hormonal markers were normal levels except plasma norepinephrine and urinary normetanephrine were modestly high (≦ 2.0-fold the upper limit of normal range). Even though the slight elevation in biochemical markers, 123I metaiodobenzylguanidine scintigraphy confirmed the abnormal uptake in the right adrenal gland, the patient was submitted to laparoscopic adrenalectomy. Surgical pathology was diagnostic of pheochromocytoma. Postoperatively, plasma cathecolamines were all in normal range and paroxysmal hypertension disappeared.
Introduction
Pheochromocytomas are rare but clinically important tumors, whose failure to diagnose can result in sudden and lethal complications [1,2]. Pheochromocytomas are usually suggested by paroxysmal symptoms such as headache, sweating, palpitation and hypertension [3]. Although sporadic pheochromocytomas are most common, there is a growing incidence of genetically driven cases possibly secondary to widespread availability of genetic testing in some parts of the world [4]. Traditionally, sporadic forms of Pheochromocytomas occur in individuals aged 40-50 years and tumors size are large [5,6]. Using improved biochemical tests and imaging modalities, they have become to be diagnosed at earlier stage, so that the size of diagnosed tumors is decreasing [6]. Here we report a case of 17-mm in the largest dimension pheochromocytoma detected by 123I -metaiodobenzylguanidine scintigraphy without significant excessive production of catecholamines. Postoperatively, her blood pressure was well controlled with half the number of antihypertensives and paroxysmal hypertension disappeared.
Case Presentation
A 77-year-old woman was referred to a local hospital for investigation of sudden and striking blood pressure elevations. She had 17-year history of hypertension well controlled with several antihypertensive agents, but she became to have recent symptom of paroxysmal hypertension. On first visit, a screening abdominal computed tomography (CT) with 1-mm thin slices revealed a 17 x 9 mm sized homogeneous mass with soft tissue density (approximately 30-40 Hounsfield units) in right adrenal gland (Figure 1). Biochemical tests revealed that plasma norepinephrine concentration was slightly elevated (824 pg/ml), however, the levels were not within the threshold range suggested for the diagnosis of pheochromocytoma (Table 1) [7]. Plasma renin activity, plasma aldosterone concentration and adrenocorticotrophic hormone were all in normal range (Table 1). Additionally, plasma cortisol was under 1.00 μg/dL after low dose (1 mg) dexamethasone overnight suppression test. Six months later, a follow-up anatomic test coupled with biochemical measurements (plasma catecholamines and urine fractionated metanephrines) were performed. An abdominal CT demonstrated that the tumor size was no change. Biochemical tests showed that plasma norepinephrine concentration (976 pg/ml) and urinary normetanephrine excretion (713 μg/gCre) were modestly high (Table 1). Although the elevation of biochemical markers for pheochromocytoma was slight, the patient still had the symptom of paroxysmal hypertension, a functional imaging was performed. The 123I-metaiodobenzylguanidine (123I-MIBG) planar image showed abnormal uptake in the right adrenal gland indicating that the tumor was a potential pheocormosytoma. Then, she was admitted to our hospital for further evaluation for possible pheocormosytoma. During testing at hospitalization, she had the paroxysmal hypertension. Plasma catecholamine levels were within normal limits, whereas 24-hour urinary excretion rates of norepinephrine, VMA and normetanephrine were higher than upper limits of normal rang, especially the urinary norepinephrine excretion elevated to more than three times the normal value (Table 1). On the basis of the symptom, the positive 123I-MIBG scintigraphy and the significant elevation of biochemical markers, the patient was subjected to laparoscopic right adrenalectomy. Postoperative pathologic findings confirmed pheochromocytoma (Figure 2). Histologically, well demarcated area was detected within the adrenal medulla. The region was composed of adrenal medullar cells with clear/eosinophilic cytoplasm arranged in zellballen pattern (Figure 2 A and B). Immunohistochemically, the tumor cells stained positive for Chromogranin A (Figure 2C). The tumor cells exhibited a low Ki-67 index (Figure 2D). Postoperatively, her plasma cathecolamine levels were all in normal range. She remained free from symptom of paroxysmal hypertension even though she still needed antihypertensive agents to control her hypertension.
Figure 1
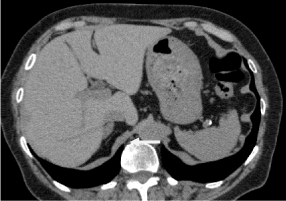
Figure 1
The patient’s standing foot posture (fpi: l=+6; r=+7).
Discussion
Pheochromocytomas are catecholamine-secreting tumors arising from chromaffin cells in the adrenal medulla. They are rare but clinically important tumors, whose failure to diagnose can result in sudden and lethal complications [1,2]. Based on autopsy studies,however, many cases are probably missed than are diagnosed. At the Mayo clinic in the 50-year period (1928-1977), 40,078 autopsies were performed and pheochromocytomas were present in 54 patients. Among these patients, 13 patients were diagnosed during life, and 41 patients were unsuspected autopsy findings [8]. On the other hand, even among patients suspected to have a pheochromocytoma, the diagnosis is rarely confirmed [9]. Because pheochromocytomas can have a highly variable presentation and an overlooked clinical entity, the diagnosis of pheochromocytomas is difficult, especially when a tumor size is small. In the present case, there are several practical points related to the diagnosis and management of small pheochromocytomas. First, small pheochromocytomas did not always have excessive catecholamine production. Marlon et al showed a direct correlation between tumor size and hormone level that was independent of the clinical presentation [5]. Run Yu et al. [6] showed that most of the patient with small pheochromocytomas exhibits modestly elevated or normal biochemical marker levels, resulting in underdiagnosis. Other study showed that small tumors have slower turnover rates and release free catecholamines into the circulation that lead to hyperadrenergic symptoms [10]. In our case, repeated biochemical measurements of plasma catecholamines and urinary catecholamines or fractionated metanephrines yield false-negative results. Plasma norepinephrine level (976 pg/ml) and urinary excretion of fractionated metanephrines (713 μg/gCre) were modestly high (plasma fractionated metanephrines are not available in Japan), however, the levels were not within the diagnostic threshold for pheochromocytoma. Therefore, referred to the recommended testing algorithms, pheochromocytoma should have been excluded in the present case [7]. Currently, there exist several guidelines based on which biochemical tests should be used to confirm or exclude a suspected pheochromocytoma, but no guideline address the importance of tumor size in the diagnosis of pheochromocytomas [11,12]. Our case suggests that it is essential to pay sufficient attention to the influence of tumor size on biochemical measurments because small pheochromocytoma often exhibits modestly elevated or normal biochemical marker levels, resulting in underdiagnosis.
Secondly, small pheochromocytoma exhibits same suspicious features on imaging as larger tumor, regardless of low biochemical marker levels [6,13]. In the present case, abdominal CT identifies 17-mm mass of which appearance of homogeneity with soft-tissue density (approximately 40-50 HU) is suggestive of a pheochromocytoma. Moreover, 123I-MIBG scintigraphy clearly showed a focus of radioactivity in the mass. The current 123I-MIBG scintigraphy has a high sensitivity at 83%-100% for a specificity of 95%-100% [14]. Owing to high sensitivity and specificity, Ioannis et al. [14], say that the presence of pheochromocytomas should always be ruled out or confirmed with I-MIBG scintigraphy. Run Yu et al. [6], also recommends to incorporate imaging characteristics of the tumor into the interpretation of biochemical test results. CT with contrast wash-out studies are alternative imaging modalities which may help characterize suspicious adrenal lesions or pheochromocytomas.
Finally, in our patient, small pheochromocytoma did not contribute to hypertension. Her hypertension didn’t extremely improve after the operation. This may be come from her long duration of hypertension and progression of arteriosclerosis. A prior study demonstrated that only a quarter of the patients with small pheochromocytomas can expect improvement of preoperative hypertension after tumor removal [6]. Regardless of a poor improvement of hypertension, resection of small pheochromocytoma is necessary, because most of tumors, if untreated, cardiovascular morbidity and mortality are high [15].
Table 1
Table 1
Biochemical testing.
Figure 2
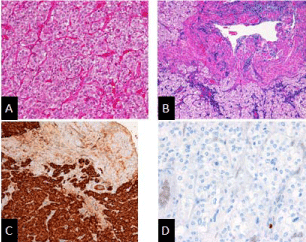
Figure 2
Pathohistological findings.
(A,B) Hematoxylin and Eosin staining. Well demarcated area was detected within the adrenal medulla The region was composed of adrenal medullar cells with clear/eosinophilic cytoplasm arranged in zellballen pattern.
(C) Immunohistochemical staining. The tumor cells stained positive for Chromogranin A.
(D) Ki-67 immunohistry. The tumor cells’ Ki-67 proliferation index was 0 %.
Conclusion
A diagnosis of probable pheochromocytoma should still be pursued in patients with modestly elevated catecholamine profiles, especially if clinical history or imaging findings are suggestive. The presence of typical adrenergic symptoms, oscillating blood pressures, known genetic predisposition or characteristic radiological findings would increase the pre-test probability of diagnosing a catecholamine producing tumor. Consideration should be given to functional imaging modalities in such cases.
References
- Whitelaw BC, Prague JK, Mustafa OG, Schulte KM, Hopkins PA, Gilbert JA, et al. Phaeochromocytoma [corrected] crisis. Clin Endocrinol (Oxf). 2014; 80: 13-22.
- Prejbisz A, Lenders JW, Eisenhofer G, Januszewicz A. Cardiovascular manifestations of phaeochromocytoma. J Hypertens. 2011; 29: 2049-2060.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005; 366: 665-675.
- Gimenez-Roqueplo AP, Lehnert H, Mannelli M, Neumann H, Opocher G, Maher ER, et al. Phaeochromocytoma, new genes and screening strategies. Clin Endocrinol (Oxf). 2006; 65: 699-705.
- Guerrero MA, Schreinemakers JM, Vriens MR, Suh I, Hwang J, Shen WT, et al. Clinical spectrum of pheochromocytoma. J Am Coll Surg. 2009; 209: 727-732.
- Run Y, Allison P, Meng W. Small Pheochromocytomas. Significance, Diagnosis, and Outcome. J Clin Hypertens. 2012; 14: 307-315.
- Study Group of “Pheochromocytoma Survey and Making Practice Guideline”, Research Enterprise of Overcoming Refractory Disease, Ministry of Health, Labour and Welfare. Clinical Guide to the Management of Pheochromocytoma 2012.
- Arora S, Vargo S, Lupetin AR. Computed tomography appearance of spontaneous adrenal hemorrhage in a pheochromocytoma. Clin Imaging. 2009; 33: 314-317.
- Fogarty J, Engel C, Russo J, Simon G, Katon W. Hypertension and pheochromocytoma testing: The association with anxiety disorders. Arch Fam Med. 1994; 3: 55-58.
- Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991; 40: 544-556.
- Goldstein DS, Eisenhofer G, Flynn JA, Wand G, Pacak K. Diagnosis and localization of pheochromocytoma. Hypertension. 2004; 43: 907-910.
- Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab. 2007; 3: 92-102.
- Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007; 356: 601-610.
- Ioannis I, Karel P. Current Approaches and Recommended Algorithm for the Diagnostic Localization of Pheochromocytoma. J Clin Endocrinol Metab. 2004; 89: 479-491.
- Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and Paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014; 99: 1915–1942.