Case Report
Cladribine for the Management of Erdheim-Chester Disease in Adults
Patel H*, Kraft C and Gary J. Schiller
Department of Hematology, University of California, Los Angeles, USA
*Corresponding author: Harsh Patel, Department of Hematology, Room 42121 Center for Health Sciences, David Geffen School of Medicine at University of California, Los Angeles, CA 90095, USA
Published: 27 Jun, 2016
Cite this article as: Patel H, Kraft C, Gary J. Cladribine for
the Management of Erdheim-Chester
Disease in Adults. Ann Clin Case Rep.
2016; 1: 1023.
Abstract
Erdheim-Chester Disease (ECD) is a rare, non-Langerhans cell histiocytosis characterized by foamy infiltrates of soft tissue and bone, with a histopathology that reveals CD68+, CD1a-, and S100- histiocytes densely infiltrating organ systems such as bone, large vessels, heart, and lungs, and other tissues to name a few. Few chemotherapeutic options exist in the second line, of which one is cladribine. Cladribine (2-chlorodeoxyadenosine) is an anti-metabolite that predominantly affects blood cells by mimicking adenosine nucleosides, inhibiting adenosine deaminase, and thus disrupts the ability of the cell to repair DNA. Here we report three patients with varying sites of disease who achieved a response following treatment with cladribine. Although the efficacy of cladribine has been demonstrated in patients with ECD who primarily exhibited neurological symptoms, we present three patients in whom significant responses were achieved in disease distributed in long bones, pericardium, and retroperitoneum.
Keywords: Cladribine; Erdheim-Chester disease; Non-Langerhans; Histiocytosis
Introduction
Erdheim-Chester disease (ECD) is a non-Langerhans cell histiocytosis that was first
described as a "lipoid granulomatous" by JakobErdheim and William Chester in 1930. The
disease is characterized by infiltration of foamy macrophages into various tissues with associated
xanthogranulomas inflammation. ECD is a relatively rare condition with between 550-600 reported
cases in the literature. Diagnosis of the disease primarily hinges on imaging, clinical symptoms, and
the aforementioned histology [1]. The disease shows preference to males and adults, although it can
present in children [2].
The pathophysiology of ECD is yet to be fully understood. There is some debate as to the reactive
or malignant nature of the disorder, with favorable data existing for both mechanisms. Stoppacciaro
et al. have shown undetectable Ki-67 on ECD histiocytes and the absence of mitosis, which led
them to conclude the limited contribution of proliferation to the disease [3]. Furthermore, they
found an increase in chemokine receptors for monocyte migration that allows for the possibility
that proliferation has a minimal contribution to the pathogenesis of the disease and an autocrine
loop of recruitment and accumulation may be the major contributing factor [4,5]. The group was
also able to show T-helper lymphocyte infiltration with IFN-γ staining with IL-10 on the histiocytes,
pointing to a TH1 or TH2-oriented inflammatory response. The alternative hypothesis, which posits
malignancy, cites the incidence of the mutation in the proto-oncogene BRAF, a member in the
MAPK pathway [6]. Depending on the study and the method used to evaluate the mutation status,
more than 50% of all tested ECD biopsies carry the V600E mutation in BRAF [7-10].
Histologic lesions are characterized by cells staining for CD68 and are CD1a-negative, indicating
that the progenitors arise from the macrophage/monocyte cell line rather than the dendritic line
characteristic of the more common Langerhans Cell Histiocytosis [1]. In addition, the clones of
ECD are S100 negative in 80% of the cases and are further differentiated from LCH by the presence
of cytoplasmic Birbeck granules in fewer than 20% of invading histiocytes [11].
The most commonly associated clinical feature of ECD is osteosclerosis of the long bones,
centered primarily in the metaphyses and diaphysis. In a large series of 59 cases, 45 had evidence
of radiologic disease such as osteosclerosis [11]. Extraskeletal manifestations of the disease are
varied, with common presentations included central diabetes insipidus, retroperitoneal fibrosis,
exophthalmos, aortic sheathing, pericardial involvement, cutaneous xanthalesmas, and neurological
involvement including parasthesias, paraplesias, and ataxia [2,11].
First-line treatment for ECD consists of either interferon-α or
pegylated interferon-α; interferon is thought to cause immunemediated
histiocyte killing and differentiation of immature histiocytes
[4]. The efficacy of interferon was debated until a study conducted
by Arnaud et al showed an increase in survival with interferon in a
53-patient cohort [2]. Furthermore, a positive correlation between
treating with escalated-dose regimen contingent on location of
disease (i.e. CNS involvement) has been established as well [12]. More
recent studies have shown that the response to IFN-α is variable and
again, dependent on the site of disease [13], leading most to cite its
lack of efficacy or poor tolerance justifying a search for an alternative
[13].
One alternative therapy is cladribine, a purine nucleoside
analogue that is selectively toxic to monocytes and lymphocytes [14].
It acts by interfering with single-stranded DNA repair and synthesis
of both resting and dividing monocytes and lymphocytes [14]. The
proposed treatment dosage for cladribine is 0.1-0.14 mg/kg per day
for five days on a twenty-eight day cycle for histiocytic diseases [15],
although the dosage is often modulated according to the severity of
the disease. Cladribine has several side effects including neutropenia,
anemia, thrombocytopenia, headaches, and increased risk of
infection, fatigue, pyrexia, optical nerve toxicity, and lymphopenia
[15,16].
Cladribine use in the management of ECD is poorly understood,
mainly due to insufficient reports. REMOVED: One of the few reports
presented a case with prominent orbital disease that was successfully
managed with cladribine treatment for two years. Beyond this report,
cladribine has been used in moderate to severe diseases; most often
in cases with CNS involvement. Induction treatment with cladribine
isn't initiated until the disease is refractory to multiple treatments,
which may contribute to the anecdotal opinions of some clinicians
against the drug. Here we present three cases of patients whose
disease was successfully managed with cladribine.
Case Presentation
Case 1: Male, Age 58
The patient presented with a history of histiocytic granulomatous
disease involving his skin, uveal tract, testis, retroperitoneum, lungs,
and diabetes insipidus (ECD), and disseminatednocardiosis. At initial
presentation to us the patient had received corticosteroids, vinblastine,
IFN-α, and mycophenolate mofetil. Despite these treatments, his
disease progressed as evidenced by intertriginous inguinal erosive
and ulcerative dermopathy and dermatologic involvement of
external auditory canals. The patient had initially been diagnosed
via testicular biopsy, which revealed granulomatous infiltration.
Upon referral to our clinic, he was treated with local radiotherapy
for intertriginous rash but was switched to IV cladribine (0.14 mg/
kg) on the aforementioned schedule due to disease uncontrolled
locally and systemically. He received a total of five cycles. There was
immediate control of the skin disease. His course was complicated
by opportunistic gram-positive disseminated nocardia infection of
the wrist, chest, and pleura during his final cycle. (Was that really
the timing? Can you provide me with a treatment synopsis?).
Since treatment over thirteen years ago, there has been persistent
retroperitoneal fibrosis, but no return of cutaneous, ocular, skeletal,
lung, or testicular manifestation of ECD since and remains clinically
stable. BRAF mutation status is unknown and the patient's disease is
followed via CT.
Case 2: Female, Age 27
A 27 year old female presented to clinic with a history of fevers,
chills, night sweats, and recently had begun experiencing weight loss
attributed to anorexia. Upon physical examination, splenomegaly
was noted. A CT at the time revealed osseous lytic bone disease,
with a subacute fracture at the posterior 10th rib and femoral neck
lesion. A subsequent CT revealed extensive bone disease, with
involvement in the right humoral shaft, proximal left radius, a
larger fracture in the 10th rib, and thickening of the abdominal
aorta and iliac arteries. Pathology was negative for both S100 and
CD1a, leading to the diagnosis of ECD after the pathologist noted
fibrosis that was inconsistent with Langerhans Cell Histiocytosis.
The patient was stated on zoledronic acid (4 mg per 28 days) for
the lytic lesions and IV cladribine (0.14 mg/kg) for management of
the disease. The patient was treated for three cycles and experienced
mild nausea and manageable leukopenia, but bone surveys revealed
decrease in lytic lesions in the right humeorous, left radius, bilateral
ribs, and was accompanied by increased sclerosis indicative of
healing. After being clinically stable for thirteen months, the patient
returned with increased bone pain. A subsequent PET showed greater
radiographic evidence of extensive disease, with recurrence of disease
in the abdominal aorta and iliac arteries. Due to prior treatment with
cladribine, the patient was started on IFN-α but had progression of
the disease and was switched back to cladribine for sixadditional
cycles. This time leukopenia was mild but the patient developed
thrombocytopenia and intermittent bruising on the hands, arms,
and legs on the final two days of each cycle. A bone marrow biopsy
and PET showed no residual abnormalities (Figure 1), but did show
signs of mild reticulin fibrosis and significant megakaryocytopenia.
The patient is once again clinically stable with no signs of progression
for over fifteen months, although she did develop renovascular
hypertension. She, and continues to be followed. BRAF mutation
status is undetermined, but under evaluation.
Case 3: Female, Age 66
This patient presented for consultation after being evaluated
for idiopathic cardiac tamponade. Before coming to our clinic, she
experienced upper respiratory symptoms that were due to pericardial
effusions diagnosed via echocardiogram. The management of her
disease prior to presentation at our clinic consisted of two pericardial
drainages, placement of a pericardial window, pericardial stripping,
empiric trail of corticosteroids, bronchodilators, and a trial of
indomethacin; none showed any improvement. The patient then
presented to our hospital with dyspnea, chronic cough, fatigue,
chronic shoulder pain, and exertional dyspnea. Pathology of the
pericardial tissue revealed infiltrates that were positive for CD68
and CD163, and negative for S100 and CD1a, leading to her ECD
diagnosis. She started her treatment with cladribine (0.14mg/kg)
that was continued for four cycles. Her course was complicated by
neutropenia attributed to the treatment, but did not result in any
significant infections. The patient noted a significant improvement
in energy, alleviation of her fatigue, and no recurrent pericardial
effusion. Her disease has been clinically stable for over four years
and follow-up ECHOs have revealed no abnormalities. Her BRAF
mutation status is undetermined.
Figure 1
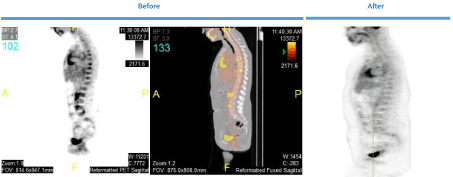
Figure 1
Patient Scans (F, 27) Figure shows the patients PET results before and after treatment with cladribine for the patient in case 2.
Discussion
Erdheim-Chester disease is a rare, non-Langerhans cell
histiocytosis marked by CD68positive, CD1anegative, and S100-
negativehistiocytes densely infiltrating bone, lymph nodes,
retroperitoneum, and systemic vasculature. Despite consensus
guidelines put in place in 2014 by Diamond et al. clinicians often
have difficulty in diagnosing ECD and therefore turn to more wellknown
treatment options for management. Fortunately multiple
drugs are now being evaluated for treating ECD, which is changing
the outlook for the disease and providing clinicians with study data
to aid their treatment decisions. ECD most commonly presents
with osteosclerosis of the long bones, centering primarily in the
metaphysis diaphysis. There is a large variance in the extra skeletal
manifestations, with common presentations in the form of diabetes
insipidus, retroperitoneal fibrosis, aortic sheathing, pericardial
involvement, and spectrum of neurological manifestations.
Despite an advantage for IFN-α in survival, it has not shown been
shown to be curative. With this in mind, more targeted therapies have
been used to treat the disease. One such drug is vemurafenib, a BRAFV600E
inhibitor, has been used to successfully treat patients with
ECD [1,13]. In the most recent study of eight patients with proven
V600E mutations showed weakened metabolic uptake as seen by PET
scan at six months, partial cardiac response in all but one patient, and
objective decreases in the size of neurological lesions when treated
with vemurafenib [6]. In one patient, treatment with vemurafenib
following IFN-α resulted in dramatic improvement of functional
capacity as well as a reduction in bone, renal, and orbital involvement
of disease [17]. Lastly, in a trial of three patients with refractory ECD
and mutated BRAF, a substantial reduction in clinical symptoms,
regression of aortic and orbital involvement and disappearance
of skin lesions was observed [13]. Other treatment methods used
include cytotoxic agents, radiation therapy, bisphosphonates,
imatinibmesylate, infliximab, anakinra, and hematopoietic stem cell
transplantation, each with variable efficacy [17].
Cladribine, the purine analog that is cytotoxic to monocytes, has
been advocated as a second-line therapy for ECD. Originally approved
for the treatment of Hairy Cell Leukemia, a hematologic malignancy
characterized by proliferation of cells from the mononuclear cell
line, it is believed that its effects may extend from plasma monocytes
to tissue histiocytes [18]. Evidence for its efficacy are scattered
throughout the literature. Cladribine has been used successfully
along with cyclophosphamide and dexamethasone to achieve partial
remissions in two patients with CNS and bone symptoms [19]. In
another case study, treatment with cladrabine resulted in remission
of orbital, pulmonary, bone, and ocular manifestations of the disease
with normalization of macrophage counts [20]. Finally, partial
remission of CNS lesions following treatment of cladribine was
consolidated with lenalidomide and resulted in a complete remission
[21].
Mutations in the proto-oncogene BRAF have been found in as few
as 50% and as many as 100% of cases, with the variability dependent
on the method of detection used. A member of the Raf kinase family,
BRAF plays a role in cell proliferation via the RAS MAPK pathway
[6]. An activating mutation, BRAF-V600E, may play a role in the
proliferation of cells derived from the mononuclear cell line [22]. The
BRAF-V600E inhibitor vemurafenib has been successfully used in
treatment of patients, most recently in a cohort of eight patients with
proven V600E mutations whose reduction in disease was measure
via reduced metabolic activity observed on PET scans [6]. Two single
case studies in which Mazor et al. [17] and Tzoulis et al. [23] noted an
excellent response in intramedullary disease, both of whom treated
with vemurafenib [23]. Lastly a study of three patients with V600E
mutated refractory ECD observed substantial reduction in clinical
symptoms, regression of aortic and orbital involvement, and the
disappearance of skin lesions when treated with vemurafenib [13].
Another targeted therapy that is gaining momentum with ECD
is sirolimus. Sirolimus is mammalian target of rapamycin (mTOR)-
inhibitor and has antiproliferative and immunosuppressive properties
[24]. If the previously presented hypothesis of ECD's pathogenesis is
correct, mTOR inhibition would manage both paths of the disease. In
a study conducted by Gianfreda and Nicastro et al ten patients were
treated with sirolimus, of whom eight were able to achieve stable
disease or an objective response with mild treatment related toxicities
[24]. Additional immunohistochemistry and immunofluorescence
done by the group on the ECD biopsies revealed mTOR pathway
activation via phosphorylated forms of mTOR and its downstream
kinase p70S6K. Other studies have also shown PIK3CA and NRAS
mutations, which are mTOR pathway activating mutations [22].
Here we presented three patients who presented in our clinic
for the management of their ECD. One of the patients presented
after extensive treatments (corticosteroids, vinblastine, IFN-α,
and mycophenolate mofetil); the other two had no other systemic
therapies. Each of the three patients was treated with cladribine (0.14
mg/kg) via IV infusion for two hours a day for five days on a twentyeight
day cycle. All patients have sustained disease regression after
treatments spanning five, nine, and four cycles respectively. All three
patients received anti-PCP, anti-viral, and anti-fungal prophylaxis
for one year after the completion of their therapy. The first and third
patient required no further treatment; patient two required further
treatment whose disease is now clinically stable.
Current trials include a Phase II study with dabrafenib and
trametinib in patients with the BRAF mutation (NCT02281760),
lenalidomide (NCT02523040), sirolimus with prednisone
(ACTRN12613001321730), tocilizumab (NCT01727206), and a
long-term outcome after vemurafenib inhibitor interruption study
(NCT02089724).
Here we have presented our experience with three ECD patients
with varying sites of disease whose disease was successfully managed
with cladribine. Despite this, there is a patient population who are
ineligible for investigational treatments and who would benefit from
an alternate therapy. It is here we suggest cladribine to be used as an
initial or early therapeutic option based on our experience with the
drug and the success our patients have had. Beyond this, it would
be interesting to see a larger study with cladribine or a study that
compares it to one of the targeted therapies.
Ethics Approval
Ethics approval was obtained from the IRB at UCLA. The
reference number for the study is 14-001051.
Ethics, Consent, and Permissions/Consent to Publish
All participants were consented via an IRB approved consent
form and were consented by Dr. Gary Schiller. They were notified
that their consent would mean that they are participating in this study
and the results would be published.
Acknowledgements
We would like to thank our patients for allowing us to present their cases.
References
- Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim-Chester disease. Rheum Dis Clin North Am. 2013; 39: 299- 311.
- Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, Wechsler B, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011; 117: 2778-2782.
- Stoppacciaro A, Ferrarini M, Salmaggi C, Colarossi C, Praderio L, Tresoldi M, et al. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim-Chester disease: implications for pathogenesis. Arthritis Rheum. 2006; 54: 4018-4022.
- Dagna L, Girlanda S, Langheim S, Rizzo N, Bozzolo EP, Sabbadini MG, et al. Erdheim-Chester disease: report on a case and new insights on its immunopathogenesis. Rheumatology (Oxford). 2010; 49: 1203-1206.
- Laman JD, Leenen PJ, Annels NE, Hogendoorn PC, Egeler RM. Langerhans-cell histiocytosis 'insight into DC biology'. Trends Immunol. 2003; 24: 190-196.
- Haroche J, Cohen-Aubart F, Emile JF, Maksud P, Drier A, Tolédano D, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF (V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015; 33: 411-418.
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010; 116: 1919-1923.
- . Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in ErdheimChester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012; 120: 2700-2703.
- Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in ErdheimChester disease. J Clin Oncol. 2013; 31: 398.
- Cangi MG, Biavasco R, Cavalli G, Grassini G, Dal-Cin E, Campochiaro C, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis, 2015; 74: 1596-1602.
- Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, Wechsler J, Brun B, Remy M, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996; 75: 157-169.
- Gadner H, Grois N, Arico M, Broadbent V, Ceci A, Jakobson A, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001; 138: 728-734.
- Haroche J, Amoura Z, Trad SG, Wechsler B, Cluzel P, Grenier PA, et al. Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum. 2006; 54: 3330-3336.
- Ceci A, de Terlizzi M, Colella R, Loiacono G, Balducci D, Surico G, et al. Langerhans cell histiocytosis in childhood: results from the Italian Cooperative AIEOP-CNR-H.X '83 study. Med Pediatr Oncol. 1993; 21: 259-264.
- Ng-Cheng-Hin B, O'Hanlon-Brown C, Alifrangis C, Waxman J. Langerhans cell histiocytosis: old disease new treatment. QJM. 2011; 104: 89-96.
- Imamura T, Sato T, Shiota Y, Kanegane H, Kudo K, Nakagawa S, et al. Outcome of pediatric patients with Langerhans cell histiocytosis treated with 2 chlorodeoxyadenosine: a nationwide survey in Japan. Int J Hematol. 2010; 91: 646-651.
- Mazor R, Manevich-Mazor M, Shoenfel Y. Strategies and treatment alternatives in the management of Erdheim-chester Disease Informa UK. 2013: 891-899.
- Saven A, Burian C. Cladribine activity in adult langerhans-cell histiocytosis. Blood. 1999; 93: 4125-4130.
- Adam Z, Szturz P, Van íč ek J, Moulis M, Pour L, Krejčí M, et al. Cladribine (2-chlorodeoxyadenosine) in frontline chemotherapy for adult Langerhans cell histiocytosis: A single-center study of seven cases. Acta Oncol. 2013; 52: 994-1001.
- Myra C, Sloper L, Tighe PJ, McIntosh RS, Stevens SE, Gregson RH, et al. Treatment of Erdheim-Chester disease with cladribine: a rational approach. Br J Ophthalmol. 2004; 88: 844-847.
- Adam Z, Szturz P, Pour L, Krejčí M, ZahradováL, Tomíška M, et al. [Cladribine is highly effective in the treatment of Langerhans cell histiocytosis and rare histiocytic disorders of the juvenile xanthogranuloma group]. Vnitr Lek. 2012; 58: 455-465.
- Emile JF, Diamond EL, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in ErdheimChester disease. Blood. 2014; 124: 3016-3019.
- Tzoulis C, Schwarzlmüller T, Gjerde IO, Søfteland E. Excellent response of intramedullary Erdheim-Chester disease to vemurafenib: a case report. BMC Res Notes. 2015; 8: 171.
- Gianfreda D, Nicastro M, Galetti M, Alberici F, Corradi D, Becchi G, et al. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015; 126: 1163-1171.